
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient dynamic asymmetric catalytic system within a zinc-templated network†‡
Anna
Serra-Pont
,
Ignacio
Alfonso
 ,
Jordi
Solà
* and
Ciril
Jimeno
*
,
Jordi
Solà
* and
Ciril
Jimeno
*
Department of Biological Chemistry, Institute of Advanced Chemistry of Catalonia (IQAC-CSIC), Jordi Girona 18-26, E08034 Barcelona, Spain. E-mail: jordi.sola@iqac.csic.es; ciril.jimeno@iqac.csic.es
First published on 13th June 2019
Abstract
Enhanced cooperativity leading to high catalytic activity and stereoselectivity has been achieved through a complex network of simple species interacting reversibly. This novel dynamic catalytic system relies on bipyridine-based organocatalytic ligands and zinc(II) as the template. It demonstrates the effectiveness of dealing with mixtures rather than single species in asymmetric catalysis.
Addressing the properties and behaviour of complex chemical networks is a challenging task, especially when the achievement of a specific function is pursued. In metal catalysis or supramolecular catalysis, several species might coexist in dynamic equilibrium, only one of them being the actual (or most active) catalyst. The identification and study of such catalysts might be problematic as well, and leads to difficult rationales and subsequent unavoidable trial-and-error optimization.
After the emergence of Systems Chemistry,1,2 these complex systems can be seen in a different manner: the generation of new catalysts is somehow circumvented by focusing on the network design instead of the preparation of a unique catalyst.
To this aim, the understanding of the catalytic network as a whole is needed, which is not trivial. As a consequence, examples of new catalytic networks are still scarce.3 Pioneering examples were disclosed by Otto and co-workers using disulfide exchange to generate new catalysts for the Diels–Alder or Cope reactions from a library of building blocks.4–6 A few other examples have been subsequently developed using alternative approaches, essentially reversible imine formation.7–11 However, none of them dealt with controlling the stereoselectivity of the catalytic reaction. Thus, there is lack of important knowledge in this field.
We recently approached this issue through a simple methodology combining our developments in dynamic systems12–19 and organocatalysis.20–23 In our approach, the rapidly exchanging metal−pyridine ligand bonds furnished the catalytic network. The dynamic system thus generated contained sufficient amounts of a catalytically (kinetically) competent self-assembled species leading to high reaction rate acceleration and subsequent high enantioselectivity in the direct aldol reaction. No particular coordination complex could be isolated. The above system was optimized on a trial-and-error basis.24,25
In this communication, we report on the design and synthesis of a new dynamic organocatalytic system of unprecedented functional ligands based on a bipyridine scaffold (bipyPro and bipyTU, Fig. 1) for the asymmetric aldol reaction. The rationale behind this system stems from the good binding ability of bipyridine to zinc, leading to slower ligand exchange and thus to a more defined and efficient catalytic network compared to the parent pyridine-based catalyst. Cooperativity emerging from this network in the form of a Zn(bipyPro)(bipyTU) complex is the key to success, which has been optimized on the basis of a complete understanding of its behaviour and equilibria.
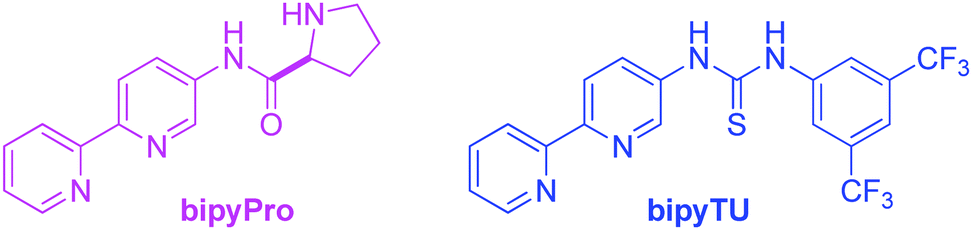 | ||
| Fig. 1 Functional bipyridine ligands. | ||
The new bipyridine-based ligands bipyPro and bipyTU (Fig. 1) were synthesized in a straightforward manner in fairly high overall yields (see ESI‡). Since the metal–ligand combination is a key parameter in these systems, we examined several zinc(II) salts in combination with the above mentioned ligands for the asymmetric aldol reaction of cyclohexanone and p-nitrobenzaldehyde. Zinc trifluoroacetate turned out to be the best match for the bipyridines, resulting in full conversion and 92% ee (entries 1–6, Table 1).26
|
|
|||||
|---|---|---|---|---|---|
| Entry | ZnX2 | Reaction time/h | Conversiona/% | anti/syna | eeb/% |
| a Determined by 1H NMR on crude samples. b Determined by HPLC on a chiral stationary phase. c Reaction run at 0 °C. d 5 mol% loading of metal and ligands. e 2 mol% loading of metal and ligands. | |||||
| 1 | ZnCl2 | 24 | 5 | 82/18 | n.d. |
| 2 | Zn(BF4)2 | 24 | 72 | 81/19 | 85 |
| 3 | Zn(OTf)2 | 24 | 67 | 81/19 | 76 |
| 4 | ZnSO4·7H2O | 24 | 8 | 76/24 | n.d. |
| 5 | Zn(OTs)2 | 24 | 84 | 81/19 | 85 |
| 6 | Zn(O2CCF3)2 | 24 | 99 | 80/20 | 92 |
| 7 | 6 | 91 | 85/15 | 91 | |
| 8c | 8c | 96c | 92/8c | 99c | |
| 9c,d | 14c,d | 89c,d | 91/9c,d | 97c,d | |
| 10c,e | 14c,e | 48c,e | 89/11c,e | 98c,e | |
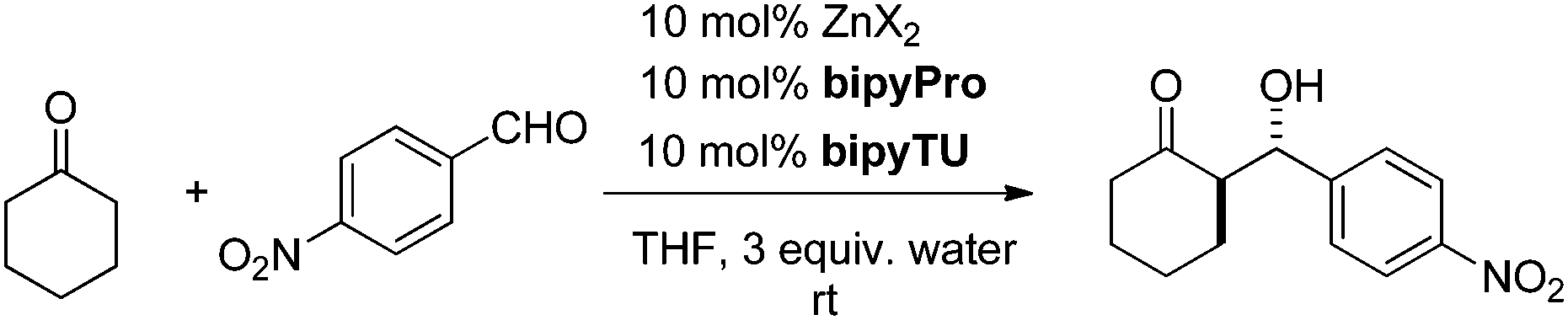
The addition of a small quantity of water (3 equiv.) was crucial to ensure the activity of the catalytic system, as we have always encountered during the development of organocatalytic Michael and aldol reactions.20–27 Moreover, our methodology involves the catalyst pre-formation in solution for 1 hour at room temperature before the reaction temperature is set and substrates are added. Otherwise, an induction period is observed before the catalytic activity can be detected (see ESI‡ for details).
The catalyst loading could be initially set to 10 mol% of metal and ligands. In this way, high conversion (>90%) and stereoselectivity (91% ee) were achieved in just 6 hours at room temperature (entry 7, Table 1). At this stage, it was only necessary to run the reaction at 0 °C, and >90% conversion and 99% ee were obtained (entry 8, Table 1). It was even possible to further reduce the amount of catalytic species down to 5 mol% by slightly increasing the reaction time and still keeping synthetically useful results. Even at 2 mol% loading, the stereoselectivity of the reaction was excellent, although the reaction rate was slower (entries 8 and 9, Table 1).
We then performed a series of NMR titrations to shed light on the nature of the catalytic system. In accordance with our expectations, a dynamic system with ligand exchange was under operation, at an intermediate rate on the 1H NMR time scale and a slow rate according to the 19F NMR time scale (see ESI‡). Unfortunately, no more precise data could be gathered from these experiments.
A deeper study of the dynamic catalytic system was then faced through two indirect but converging approaches: (1) a UV-Vis study of the metal–ligand mixture in order to determine the equilibrium constants and a model of formation of the catalyst, and (2) a conversion evolution with time under different catalytic conditions to infer the nature of the catalytic species and the increase of cooperative effects.
First, we envisioned the otherwise reasonable equilibrium network model in Scheme 1, in which several species compete for the reaction or are ineffective, while only one is able to generate cooperative catalysis.28 In this model, all the bipyridine-metal species are in equilibrium, in accordance with our 1H NMR observations. Complexes with up to two bipyridine ligands can be formed, and ligand exchange between these saturated complexes must take place through a dissociative mechanism. The model was then validated through a series of UV-Vis titrations of bipyPro, bipyTU, and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of bipyPro and bipyTU with zinc(II) trifluoroacetate. The final titration experiment spectra are shown in Fig. 2. It can be clearly seen how the unbound bipyridines main band at 302 nm decreases while two new bands grow at 224 and 332 nm. This change is due to the conformational flipping between the free and bound forms of bipyridines (Fig. 2). In this way, the equilibrium constants KMP, KMP2, KMT and KMT2 were independently determined and used in the overall model containing all the species. A nice fitting to the model proposed in Scheme 1 was achieved (see ESI‡). The equilibrium constants found are shown in Fig. 2. The mixed complex Zn(bipyPro)(bipyTU) depicts the highest cumulative formation constant (KMPKMPT = KMTKMTP = 34.7 × 1010 M−2vs. KMPKMP2 = 4.2 × 1010 M−2 and KMTKMT2 = 11.2 × 1010 M−2). This is indeed an unpredictable but favorable result that drives the equilibria involved in the catalytic network towards the desired species.
:1 mixture of bipyPro and bipyTU with zinc(II) trifluoroacetate. The final titration experiment spectra are shown in Fig. 2. It can be clearly seen how the unbound bipyridines main band at 302 nm decreases while two new bands grow at 224 and 332 nm. This change is due to the conformational flipping between the free and bound forms of bipyridines (Fig. 2). In this way, the equilibrium constants KMP, KMP2, KMT and KMT2 were independently determined and used in the overall model containing all the species. A nice fitting to the model proposed in Scheme 1 was achieved (see ESI‡). The equilibrium constants found are shown in Fig. 2. The mixed complex Zn(bipyPro)(bipyTU) depicts the highest cumulative formation constant (KMPKMPT = KMTKMTP = 34.7 × 1010 M−2vs. KMPKMP2 = 4.2 × 1010 M−2 and KMTKMT2 = 11.2 × 1010 M−2). This is indeed an unpredictable but favorable result that drives the equilibria involved in the catalytic network towards the desired species.
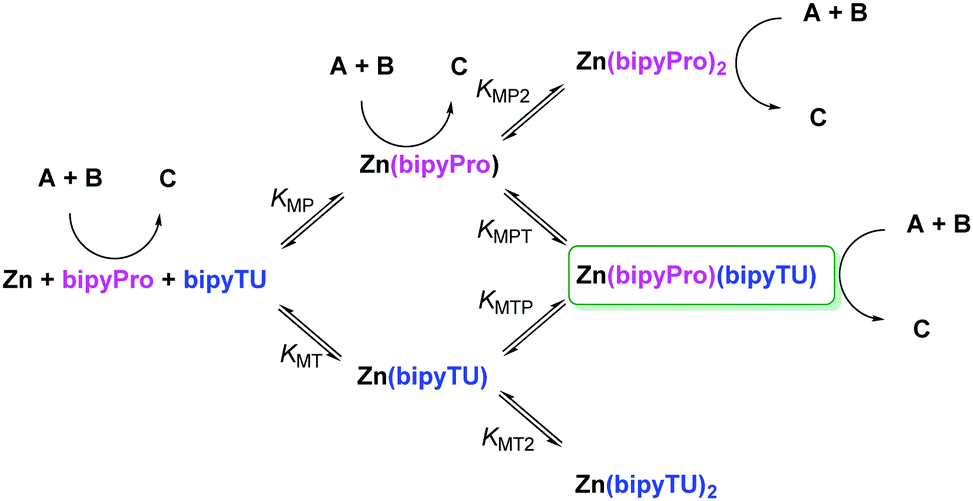 | ||
| Scheme 1 Model for the Zn(O2CCF3)2–bipyPro–bipyTU system in equilibrium, highlighting the potential catalytic species. K represents the equilibrium constants. Subscript M stands for Zn(O2CCF3)2, P for bipyPro and T for bipyTU. | ||
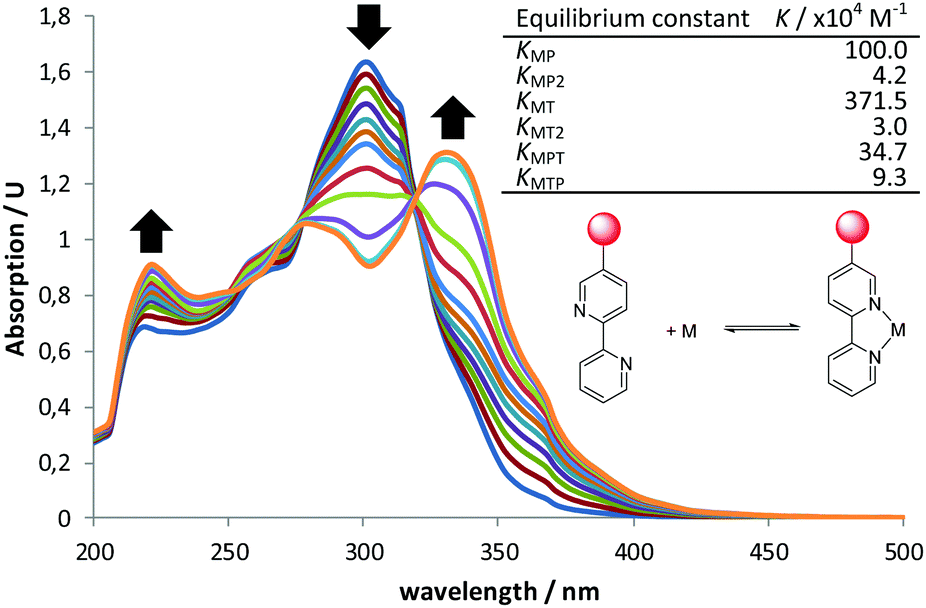 | ||
| Fig. 2 UV-Vis titration at rt of 1:1 bipyPro:bipyTU with Zn(O2CCF3)2 and calculated equilibrium constants for the system model in Scheme 1. Cumulative β constants fitting and error can be found in the ESI.‡ | ||
Next, the asymmetric aldol reaction was carried out with different catalytic components and conversion vs. time plots were constructed (Fig. 3). Poor turnover and moderate ee's were observed with free bipyPro or a combination of free bipyPro and bipyTU. We did observe significant rate acceleration when zinc(II) trifluoroacetate and bipyPro were combined, likely due to a prolinamide activation by Lewis acid.29–31 A moderate 83% ee in the aldol product was obtained in that case. Most importantly, we determined a high rate acceleration and high enantioselectivity when the full catalytic system (Zn salt–bipyPro and bipyTU) was used. Indeed, a 3-fold rate increase was determined due to the sole addition of the thiourea ligand, showing the emergence of cooperativity with the three catalytic components (see Fig. 3 and ESI‡), and suggesting the formation of a metal-templated bifunctional species.32,33
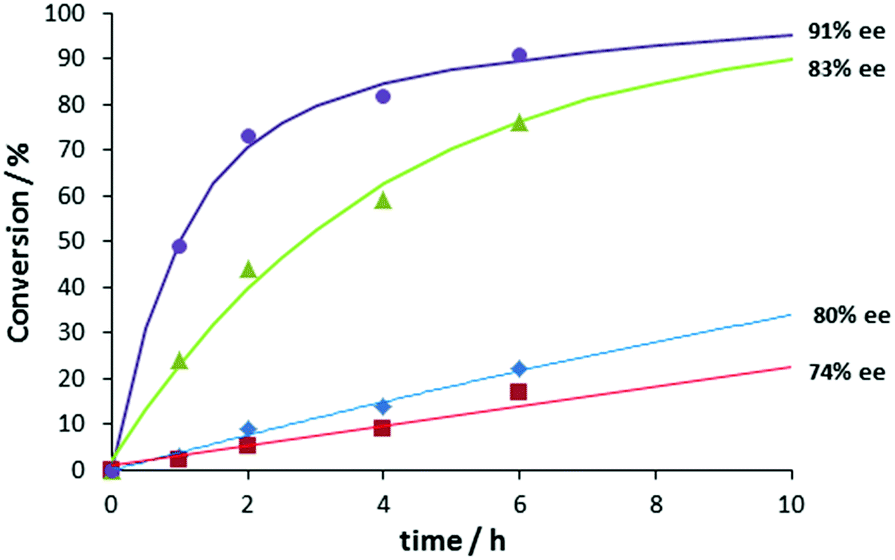 | ||
| Fig. 3 Comparison of activity and enantioselectivity of different components in the catalytic system, at room temperature and 10 mol% catalyst loading. bipyPro, blue diamonds; bipyPro + bipyTU, red squares; Zn(O2CCF3)2 + bipyPro, green triangles; Zn(O2CCF3)2 + bipyPro + bipyTU, purple circles. | ||
Further proof of a single species as the major catalyst involving the three components comes from kinetic evidence: the reaction is first order in the catalyst. Moreover, the absence of non-linear effects (NLE) also suggests a single, predominant catalytic species (see ESI‡ for details).
The results in Fig. 3 are also important because they tell us that the competitive reaction catalyzed by free bipyPro in Scheme 1 is of minor importance (ca. 30% conversion and 80% ee maximum after 10 hours). However, the potential competitive effect of the Zn–bipyPro combination leading to diminished enantioselectivity (>80% conversion and 83% ee, Fig. 3) was obviously not negligible a priori. We thus analyzed the relative concentrations of catalytic species under our conditions using the above model. The maximum concentration of the most active Zn(bipyPro)(bipyTU) complex actually occurs under the reaction conditions ([Zn(O2CCF3)2] = [bipyPro] = [bipyTU] = 0.06 M): 71% of all the species in the medium correspond to the bifunctional complex, whereas 14% correspond to the less efficient species Zn(bipyPro)2. The amounts of free bipyPro or Zn(bipyPro) are negligible (see ESI‡).
For further security, the reaction was tested at 10 mol% Zn(O2CF3)2 and 20 mol% bipyPro, which should lead to the formation of Zn(biyPro)2 as the major component of the system. The reaction was slower and less enantioselective (72% ee anti) compared to the full catalytic system that actually contains half the amount of chiral ligand (see ESI‡). Altogether, these results point out that Zn(bipyPro)(bipyTU) is the fundamental catalyst of the network.
Therefore, we propose a model that explains the catalytic activity and stereoselectivity of the reaction. A tetrahedral zinc complex would be able to form the enamine through the prolinamide moiety, whereas the thiourea group from the perpendicular ligand would approach the aldehyde through hydrogen bonding in a way that steric repulsion from the inner complex is minimized (Fig. 4).
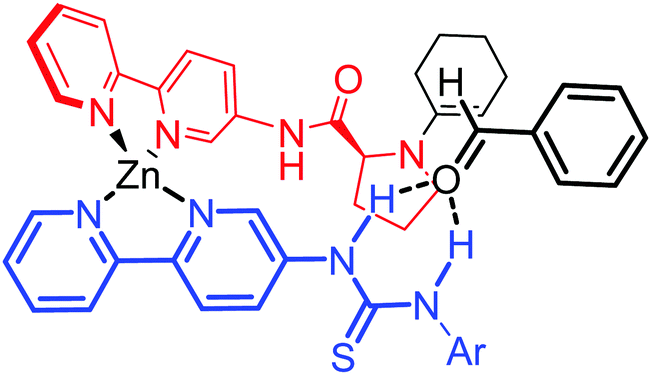 | ||
| Fig. 4 Stereoselectivity model for the asymmetric aldol reaction with Zn(bipyPro)(bipyTU) as the catalyst. | ||
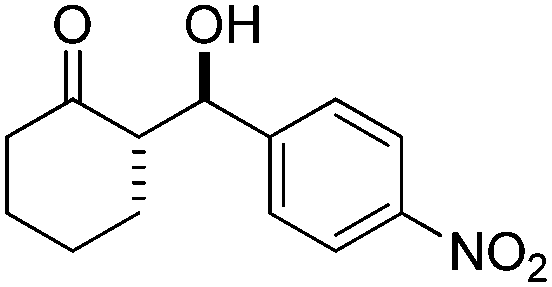
The scope of this catalytic system was tested with different substrates (Table 2). Excellent results in both yield and stereoselectivity were obtained with just 5 mol% catalyst loading when cyclohexanone and strong electron withdrawing aldehydes were used (p-NO2 and p-CF3 benzaldehyde). In these cases, the ee climbed up to 99% with 10 mol% catalyst (Table 2, entries 1 and 2). Other substituted benzaldehydes with electron withdrawing groups also performed very well, as well as the 1,4-cyclohexandione monoethylene acetal with p-nitrobenzaldehyde (entries 3–6). Unfortunately, the reaction of cyclohexanone with benzaldehyde was sluggish, although with excellent enantioselectivity (entry 7). This is not strange since we and others already showed some time ago the important electronic effects that hamper the reactivity of electron rich aldehydes in this reaction.23,34–36 In turn, cyclopentanone and acetone yielded the aldol products in high yields but modest stereoselectivities (entries 8 and 9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Product | Catalyst loading/mol% | Yielda/% | d.r.banti/syn | eecanti/% |
| Reactions run for 6–14 h.a Isolated yield after flash chromatography.b Determined by 1H NMR.c Determined by HPLC on a chiral stationary phase.d Not isolated. | |||||
| 1 |
|
5 | 87 | 90/10 | 95 |
| 10 | 90 | 92/8 | 99 | ||
| 2 |

|
5 | 70 | 99/1 | 92 |
| 10 | 92 | 94/6 | 99 | ||
| 3 |
|
10 | 74 | 99/1 | 94 |
| 4 |

|
10 | 95 | 93/7 | 96 |
| 5 |
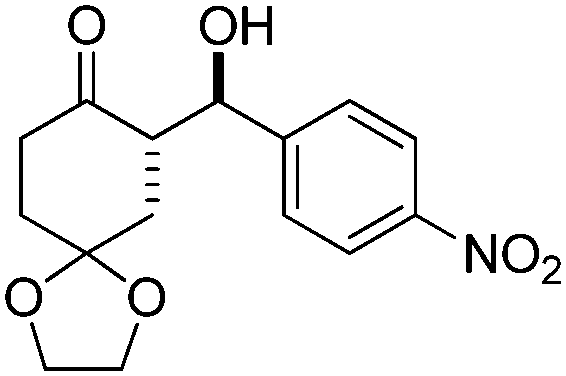
|
10 | 62 | 89/11 | 93 |
| 6 |

|
10 | 70 | 90/10 | 97 |
| 7 |
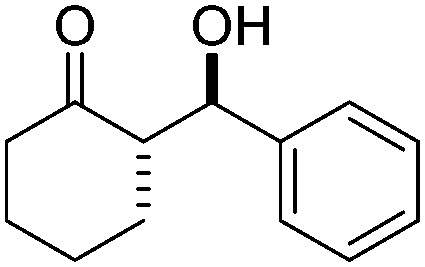
|
10 | 36d | 92/8 | 96 |
| 8 |
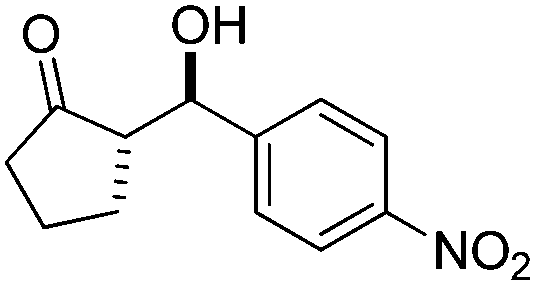
|
10 | 92 | 24/76 | 58/11 |
| 9 |
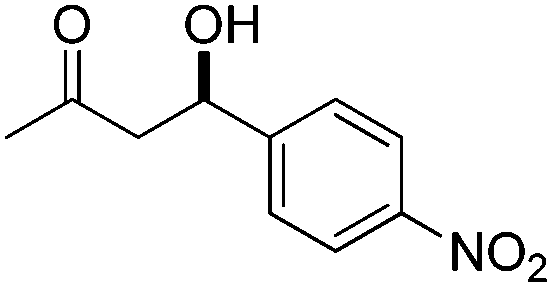
|
10 | 89 | — | 37 |
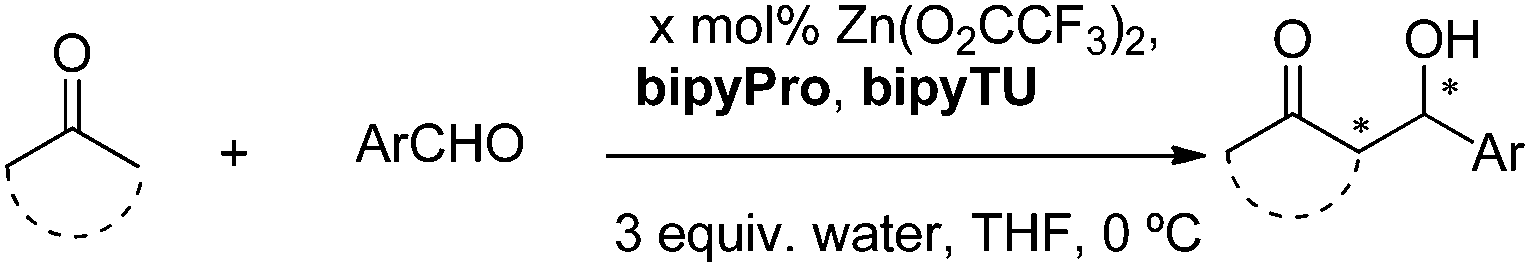
In conclusion, a novel organocatalytic dynamic system based on bipyridine ligands and zinc trifluoroacetate as the templating agent has been developed. It presents a significantly high performance in terms of reaction rate and stereoselectivity at low catalyst loading, showing efficiency attainable with such catalytic systems.37 Most importantly, the dynamic system has been analysed and rationalized in detail for the first time. As a consequence, the equilibria network of all the species present could be modelled, showing that the bifunctional, cooperative self-assembled catalyst is the predominant species under the reaction conditions. These conditions are indeed optimal to obtain the highest concentration of the desired catalytic complex. Nevertheless, it must be emphasized that, as experimental evidence points out, not only the major species but different species within the dynamic network are able to promote the reaction.
Accordingly, the observed activity (and selectivity) is a result of the network as a whole. This fact allows envisioning the opportunity for catalytic activity regulation by tuning the properties of the network. Efforts towards that direction are underway in our lab.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC.
- O. Š. Miljanić, Chem, 2017, 2, 502–524 Search PubMed.
- P. Dydio, P.-A. R. Breuil and J. N. H. Reek, Isr. J. Chem., 2013, 53, 61–74 CrossRef CAS.
- B. Brisig, J. K. M. Sanders and S. Otto, Angew. Chem., Int. Ed., 2003, 42, 1270–1273 CrossRef CAS PubMed.
- H. Fanlo-Virgos, A.-N. R. Alba, S. Hamieh, M. Colomb-Delsuc and S. Otto, Angew. Chem., Int. Ed., 2014, 53, 11346–11350 CrossRef CAS PubMed.
- L. Vial, J. K. M. Sanders and S. Otto, New J. Chem., 2005, 29, 1001–1003 RSC.
- F. Schaufelberger and O. Ramstrom, Chem. – Eur. J., 2015, 21, 12735–12740 CrossRef CAS PubMed.
- F. Schaufelberger and O. Ramstrom, J. Am. Chem. Soc., 2016, 138, 7836–7839 CrossRef CAS PubMed.
- M. Matsumoto, D. Estes and K. M. Nicholas, Eur. J. Inorg. Chem., 2010, 1847–1852 CrossRef CAS.
- G. Gasparini, L. J. Prins and P. Scrimin, Angew. Chem., Int. Ed., 2008, 47, 2475–2479 CrossRef CAS PubMed.
- G. Gasparini, B. Vitorge, P. Scrimin, D. Jeannerat and L. J. Prins, Chem. Commun., 2008, 3034–3036 RSC.
- J. Atcher and I. Alfonso, RSC Adv., 2013, 3, 25605–25608 RSC.
- J. Atcher, A. Moure and I. Alfonso, Chem. Commun., 2013, 49, 487–489 RSC.
- J. Atcher, A. Moure, J. Bujons and I. Alfonso, Chem. – Eur. J., 2015, 21, 6869–6878 CrossRef CAS PubMed.
- M. Lafuente, J. Atcher, J. Solà and I. Alfonso, Chem. – Eur. J., 2015, 21, 17002–17009 CrossRef CAS PubMed.
- J. Solà, M. Lafuente, J. Atcher and I. Alfonso, Chem. Commun., 2014, 50, 4564–4566 RSC.
- A. M. Valdivielso, F. Puig-Castellví, J. Atcher, J. Solà, R. Tauler and I. Alfonso, Chem. – Eur. J., 2017, 23, 10789–10799 CrossRef CAS PubMed.
- M. Lafuente, J. Solà and I. Alfonso, Angew. Chem., Int. Ed., 2018, 57, 8421–8424 CrossRef CAS PubMed.
- M. Corredor, D. Carbajo, C. Domingo, Y. Pérez, J. Bujons, A. Messeguer and I. Alfonso, Angew. Chem., Int. Ed., 2018, 57, 11973–11977 CrossRef CAS PubMed.
- Z. I. Günler, I. Alfonso, C. Jimeno and M. A. Pericàs, Synthesis, 2017, 319–325 Search PubMed.
- Z. I. Günler, X. Companyó, I. Alfonso, J. Burés, C. Jimeno and M. A. Pericàs, Chem. Commun., 2016, 52, 6821–6824 RSC.
- C. Jimeno, L. Cao and P. Renaud, J. Org. Chem., 2016, 81, 1251–1255 CrossRef CAS PubMed.
- A. M. Valdivielso, A. Catot, I. Alfonso and C. Jimeno, RSC Adv., 2015, 5, 62331–62335 RSC.
- A. Serra-Pont, I. Alfonso, C. Jimeno and J. Solà, Chem. Commun., 2015, 51, 17386–17389 RSC.
- A. Serra-Pont, I. Alfonso, J. Solà and C. Jimeno, Org. Biomol. Chem., 2017, 15, 6584–6591 RSC.
- The exact role of the counterion remains unknown.
- C. Jimeno, Org. Biomol. Chem., 2016, 14, 6147–6164 RSC.
- N. Singh and B. Escuder, Chem. – Eur. J., 2017, 23, 9946–9951 CrossRef CAS PubMed.
- Y. Deng, S. Kumar and H. Wang, Chem. Commun., 2014, 50, 4272–4284 RSC.
- A. Gualandi, L. Mengozzi, C. M. Wilson and P. G. Cozzi, Chem. – Asian J., 2014, 9, 984–995 CrossRef CAS PubMed.
- Z. Du and Z. Shao, Chem. Soc. Rev., 2013, 42, 1337–1378 RSC.
- L. Gong, L.-A. Chen and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10868–10874 CrossRef CAS PubMed.
- L. Zhang and E. Meggers, Chem. – Asian J., 2017, 12, 2335–2342 CrossRef CAS PubMed.
- S. Guizzetti, M. Benaglia, L. Raimondi and G. Celentano, Org. Lett., 2007, 9, 1247–1250 CrossRef CAS PubMed.
- F. Chen, S. Huang, H. Zhang, F. Liu and Y. Peng, Tetrahedron, 2008, 64, 9585–9591 CrossRef CAS.
- I. Triandafillidi, A. Bisticha, E. Voutyritsa, G. Galiatsatou and C. G. Kokotos, Tetrahedron, 2015, 71, 932–940 CrossRef CAS.
- As far as we are aware, only one example of an (isolated) metal-templated catalyst for bifunctional enamine-hydrogen bonding catalysis is known. See: H. Huo, C. Fu, C. Wang, K. Harmsa and E. Meggers, Chem. Commun., 2014, 50, 10409–10411 RSC.
Footnotes |
| † Dedicated to Prof. Philippe Renaud on his 60th anniversary. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and NMR spectra. Full titrations and model fitting, MS and kinetic data. See DOI: 10.1039/c9cc03958a |
| This journal is © The Royal Society of Chemistry 2019 |