
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWhich future for stereogenic phosphorus? Lessons from P* pincer complexes of iron(II)
Raffael
Huber
 ,
Alessandro
Passera
and
Antonio
Mezzetti
*
,
Alessandro
Passera
and
Antonio
Mezzetti
*
Department of Chemistry and Applied Biosciences, ETH Zürich, Wolfgang-Pauli-Str. 10, CH-8093 Zürich, Switzerland. E-mail: mezzetti@inorg.chem.ethz.ch
First published on 2nd July 2019
Abstract
PNP pincer ligands stabilise diamagnetic base metal catalysts, and much effort has been invested in the development of chiral analogues for asymmetric catalysis. Starting from the conformational issues that affect P-stereogenic diphosphines, we extend the analysis to our recently prepared P-stereogenic PNP pincer ligands, which we used in the iron(II)-catalysed hydrogenation of ketones. Backbone rigidity and size (and shape) of the substituents at phosphorus are pivotal in both bi- and tridentate P-stereogenic ligands, and their interplay is discussed, as well as the contribution (and shortcomings) of DFT calculations to the understanding of the conformational flexibility and enantiodiscrimination.
 Raffael Huber | Raffael Huber, born 1992 in Altdorf (Switzerland) studied chemistry at the ETH Zürich. After his Master degree in 2014 with Prof. Antonio Mezzetti on NPPN iron(II) complexes, he obtained his PhD degree in the same group in 2018 with a thesis on P-stereogenic pincer ligands in iron-catalysed reactions. |
 Alessandro Passera | Alessandro Passera was born in Magenta (Italy) in 1992. After a MSc thesis co-supervised by Prof. Anna Iuliano (University of Pisa, Italy) and Prof. Vincenzo Passarelli (University of Zaragoza, Spain), Alessandro Passera concluded his Chemistry study with the Diploma of Sciences at the Scuola Normale Superiore in Pisa. He is now PhD student at ETH Zürich under the supervision of Prof. Antonio Mezzetti. His current research is focused on Mn(I) and Fe(II) catalysts for the asymmetric hydrogenation of ketones. |
 Antonio Mezzetti | Antonio Mezzetti was born in Trieste (Italy) in 1958, where he completed his studies in chemistry. In 1986, he joined the Chemistry Department of the University of Udine as Research Associate. After sabbatical stays at the ETH Zurich with Prof. Giambattista Consiglio and the late Prof. Luigi M. Venanzi, he joined the Laboratory of Inorganic Chemistry of the ETH Zürich in 1995, where he is now Titulary Professor. His research interests are at the interface between coordination chemistry and homogeneous catalysis. |
Iron(II) and stereogenic phosphorus: the context
Scope and goal
Pincer ligands form robust complexes with base metals and have hence played a pivotal role in the development of base metal catalysts. In these studies, a handful of C-stereogenic pincer ligands have been developed prior to our study. Stereogenic phosphorus (P*) has met large success in combination with bidentate ligands,1 but had not been used with tridentate ligands for base metal catalysts. Our group has prepared P-stereogenic PNP pincer ligands and used them in the iron-catalysed asymmetric hydrogenation of ketones.The goal of this article is to put the conformational issues encountered during our study in a broader context that encompasses P-stereogenic diphosphines and (the few) PNP/PCP pincer catalysts of other metals, both precious and non-noble.
The new Frontier: chiral ligands for base metal catalysts
The first iron(II) catalysts for the asymmetric hydrogenation of polar double bonds have been designed by analogy with their ruthenium(II) analogues.7 Thus, the first asymmetric iron(II) catalyst for the hydrogenation of ketones, reported by Morris in 2008,8 was based on the tetradentate, open-chain PNNP ligand already used with ruthenium and exploited in hydrogen transfer from 2-propanol.7b It should be noted, however, that the analogy between iron(II) and ruthenium(II) is deceiving, as iron(II) complexes are considerably less stable than their heavier counterparts. However, the combination of good σ donors, such as phosphines, with strong π-accepting ancillary ligands, e.g. CO, stabilises the low spin d6 configuration in which all M–L σ* antibonding orbitals are empty. Hence, ligand lability can be greatly reduced, and robust complexes are obtained.
For instance, Morris’ second-generation PNNP catalyst ligand is stable, but yet active and achieves turnover frequencies of up to 200 s−1.9 With the same goal, our group has developed macrocyclic N2P2 ligands that, combined with isonitriles as ancillary ligands, form robust, highly enantioselective catalysts for the transfer hydrogenation of a broad scope of ketones, including a hydride complex that catalyses the base-free reduction of benzils.10
As already observed for ruthenium(II),11 the complexes of tetradentate N2P2 ligands perform well in transfer hydrogenation, but are poor catalysts for the hydrogenation with H2,12 which is preferable for industrial application due to its irreversibility and atom economy.
 | ||
| Scheme 1 Achiral Fe/PNP catalysts for direct hydrogenation. | ||
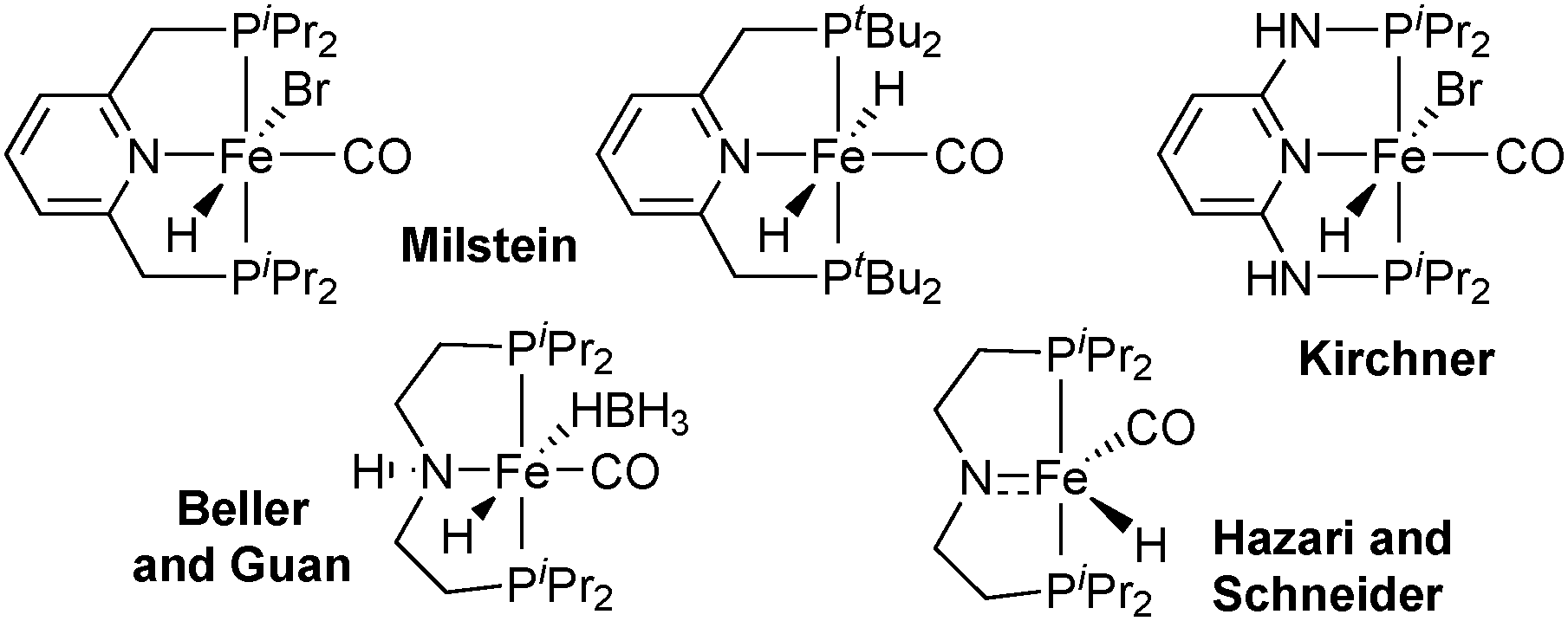
As an alternative to the pyridine backbone, Beller21 and Guan22 have used a N,N-diethyleneamine bridge for PN(H)P pincer ligands whose iron borohydridocarbonylhydride complex catalyses the hydrogenation of esters (Scheme 1). The secondary amine in the backbone directs and activates the carbonyl group towards hydride attack in the bifunctional mechanism.23 Jones extended the use of this catalyst to the acceptorless dehydrogenation and hydrogenation of N-heterocycles,24 and Hazari and Schneider reported a five-coordinate iron carbonylhydride that catalyses the acceptorless dehydrogenation of alcohols as well as ketone hydrogenation.25
All PNP pincer complexes described so far were achiral. In 2014, Morris reported the first iron(II) catalyst for the asymmetric pressure hydrogenation of ketones, a PNP bromodicarbonyl complex with an enantiopure 1-methyl-2-phenylethylene chelate that hydrogenates acetophenone with 80% ee (Scheme 2).26
 | ||
| Scheme 2 Iron catalysts for enantioselective direct hydrogenation. | ||
A second-generation chlorocarbonylhydride complex reduces acetophenone with 95% ee.27 This catalyst also reduces the heterocyclic 2-acetylfuran with full conversion and 95% ee. The more acidic 3,5-bis(trifluoromethyl)acetophenone and also the bulkier cyclohexyl phenyl ketone are not tolerated, however, and lead to low conversion (8% and 38%, respectively). Zirakzadeh introduced ferrocenyl as an additional stereogenic element, and the corresponding bromocarbonylhydride complex is an active catalyst for the hydrogenation of arylalkyl ketones (81% ee for acetophenone, Scheme 2).28
The catalysts shown in Scheme 2 all contain C-stereogenic ligands and have achieved notable enantioselectivity in selected instances, but their substrate scope is still narrow. In the quest for improvement, we have developed iron(II) catalysts with P-stereogenic pincer ligands and either pyridine (PNP) or N,N-diethyleneamine (PN(H)P) backbones. For this study, we chose dialkylphosphino units by analogy with the highly successful achiral iron(II) catalysts with pincer ligands.
An inherent difficulty of pyridine-based pincers (such as in Scheme 1 top) is that the benzylic position of the backbone is most likely to undergo deprotonation during catalysis, and a stereocentre in this position would be quickly epimerised. Hence, P-based stereocentres seemed particularly suitable. The lessons we learnt from these new PNP pincer ligands reminded us of the conformational issues that are also found in P-stereogenic diphosphines, which are recapitulated in the next section, starting with a detour to ligands with carbon based stereocentres.
P* diphosphines: conformational issues
Introduction
Restricted conformational ligand flexibility has long been considered as the key factor to achieve high enantioselectivity in catalysis, and many rigid structures from the chiral pool have been used as backbones.29 The introduction of a chelate ring between two phosphine donors is the first step toward rigidifying the system. However, chelate rings are intrinsically flexible. The classical approach to lock the conformation of the chelate ring is to introduce substituents into the P–P chelate to give C-stereogenic diphosphines. To understand the fundamental difference between P- and C-stereogenic ligands, it is necessary to examine the factors that steer the conformation of C-stereogenic ligands.Five-membered chelate rings
For diphosphine ligands that contain PPh2 donors, it has been recognized early that enantioselection derives from the chiral conformation of the phenyl rings, which, in the case of C-stereogenic diphosphines, is dictated by the stereocentres in the chelate.30 The absolute configuration of five-membered rings (λ or δ) is controlled by the preference of the (larger) substituents for equatorial positions (Scheme 3, the R′ substituent is given the third priority in the CIP rules).31 Therefore, the backbones of diphosphines with R stereocentres typically lead to a λ conformation, and S configured ones give δ conformations.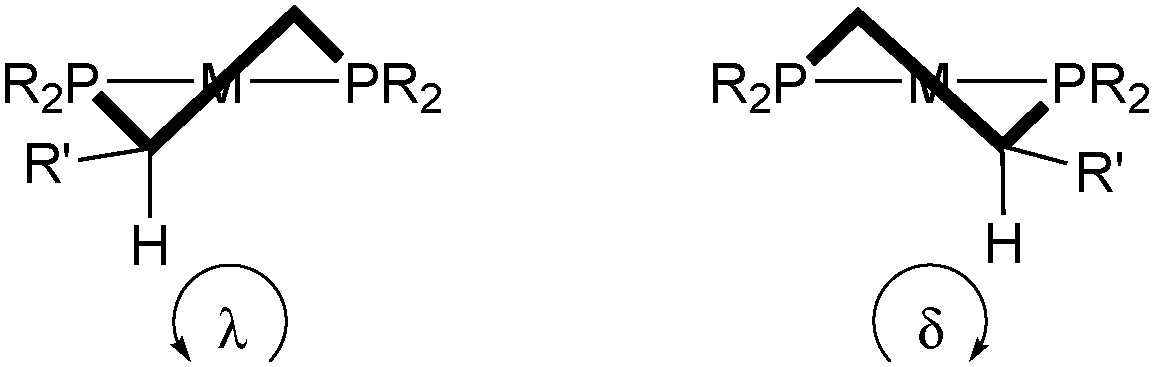 | ||
| Scheme 3 Illustration of λ (left) and δ (right) conformations in five-membered chelate rings. | ||
In turn, the conformation of the chelate determines the orientation of the phenyl rings, which transmit the chiral information to the metal-bound substrate, leading to enantioselection. In Prophos and Chiraphos, an analysis of several crystal structures indicates that the axial phenyl is preferentially edge exposed and hence interacts with the substrate more strongly than the equatorial phenyl, which tends to be face exposed (Scheme 4 top).32 However, there is a certain degree of conformational flexibility, and three types of conformations have been observed for diphosphines that form five-membered chelates such as Chiraphos (Scheme 4).32
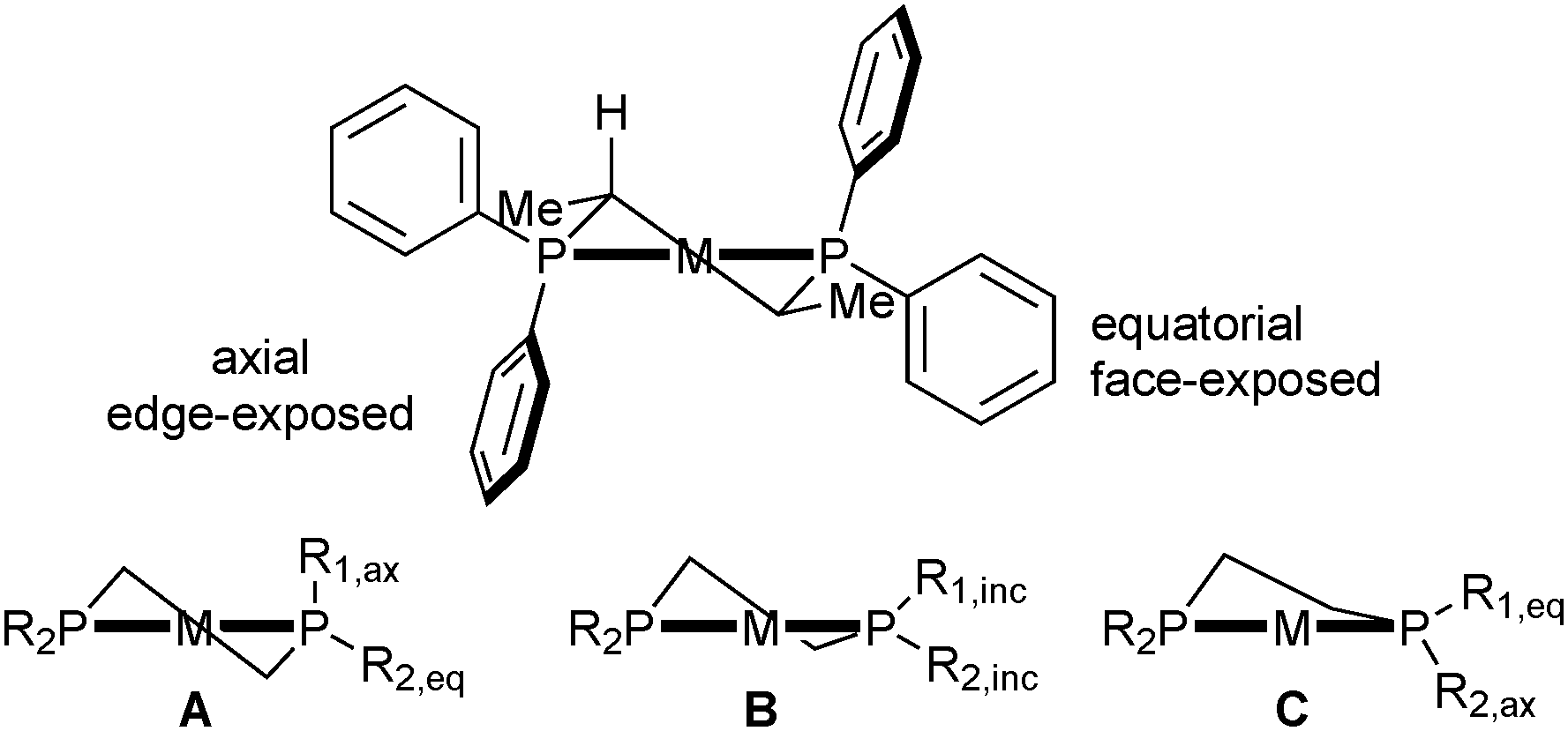 | ||
| Scheme 4 Chelate and phenyl conformation in Chiraphos. | ||
In the half-chair conformation (A), the substituents on phosphorus are clearly distinguishable as equatorial and axial and are arranged in an approximately C2-symmetric fashion. In structures close to an envelope conformation (B), which have a small P–M–P–Cring torsion angle α, the substituents on the in-plane P atom are best described as inclinal (Rinc in Scheme 4). In further distorted envelope conformations (C), the sign of the torsion angle α is inverted, and the axial and equatorial positions of the phosphorus substituents are switched with respect to A. A correlation between axial/equatorial and edge/face-exposed is possible only in half-chair structure A or in envelope structure C with a large torsion angle α (P–M–P–Cring).32
P-Stereogenic diphosphines with small chelates
The interest in P-stereogenic ligands was triggered by Knowles’ hydrogenation of α-acylaminocinnamates catalysed by Rh/DiPAMP (Scheme 5).33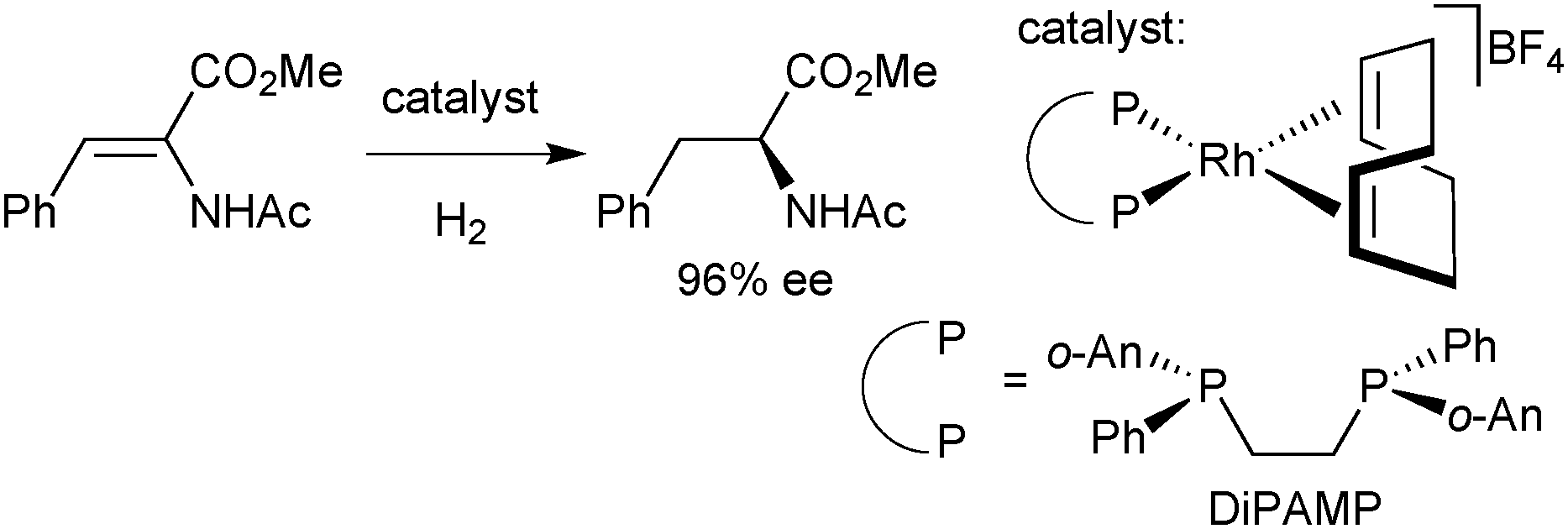 | ||
| Scheme 5 Asymmetric hydrogenation of methyl-(Z)-α-acetamidocinnamate with Rh/DiPAMP. | ||
The mechanistic investigations of enamide hydrogenation have revealed intricacies that are still a matter of debate,34,35 but have also showed that the conformation of the chelate ring is significantly more flexible in DiPAMP than in Chiraphos.36 Also, one should keep in mind that the roles of the backbone and of the phosphorus substituents are reversed in P-stereogenic diphosphines as compared to C-stereogenic ligands with PPh2 donors such as Chiraphos. Indeed, in P-stereogenic ligands, the substituents at phosphorus play a dual role as they generate the chiral environment for the approach of the substrate and lock the conformation of the chelate ring according to their size. This can lead to some confusion. Thus, in DiPAMP, the o-anisyl group acts as the “large” substituent in the chelate and hence occupies its equatorial position, but is “small” with respect to the substrate, as it is face exposed (Scheme 6). In contrast, the smaller phenyl group is axial, but edge exposed toward the substrate, and thus acts as the bulky group toward the metal-bound substrate.35 With the so-called quadrant rule, Knowles has attempted to correlate empirically the spatial arrangement of the substituents on P in the catalyst precursor (usually a diolefin complex) with the absolute configuration of the protected amino acid obtained from enamide hydrogenation.37
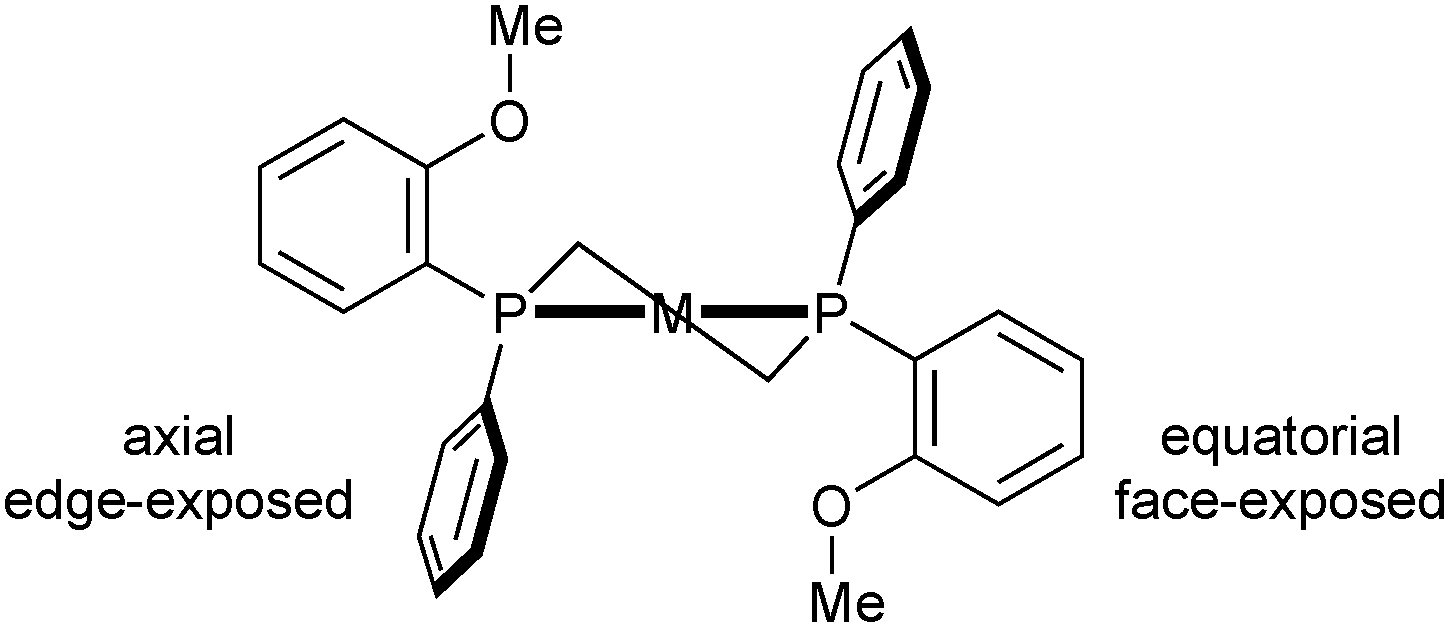 | ||
| Scheme 6 Chelate and phenyl/o-anisyl conformation in DiPAMP. | ||
However, the overall crowding in the complex affects the subtle interplay between the conformation of the chelate and the orientation of the substituents on phosphorus. Thus, the same P-stereogenic phosphine assumes opposite conformations in its norbornadiene and 1,5-cyclooctadiene complexes, but gives the same sense of induction in the hydrogenation of dehydro amino acids.38
Moreover, polysubstituted aryl substituents on phosphorus favour envelope conformations such as B and C in Scheme 4, which is beneficial for enantioselectivity, but hinders the development of simple rules-of-thumb.39 The conformational issue is acute also when the size of the P-substituents is similar, as in the case of (S,S)-tBu(Ph)PCH2CH2P(tBu)Ph,40 whose complexes show a flattened conformation of the chelate ring with no clear axial/equatorial distinction of the tert-butyl and phenyl groups. Accordingly, the tBu-substituted ligand gives the lowest enantioselectivity in the series R(Ph)PCH2CH2P(R)Ph (R = tBu, o-anisyl, 1-naphthyl).40
Therefore, the substituents on stereogenic P atoms should be significantly different in size. The P(tBu)Me unit meets this requirement, as the tert-butyl group has cylindrical symmetry with respect to P–tBu rotation and is always larger as compared to the methyl group. Imamoto's ethane-1,2-diyl-bridged, P-stereogenic diphosphines BisP* with alkylmethyl- or arylmethylphosphine donors (Scheme 7) are a privileged class of ligands and provide excellent enantioselectivity with a wide variety of catalytic reactions.41
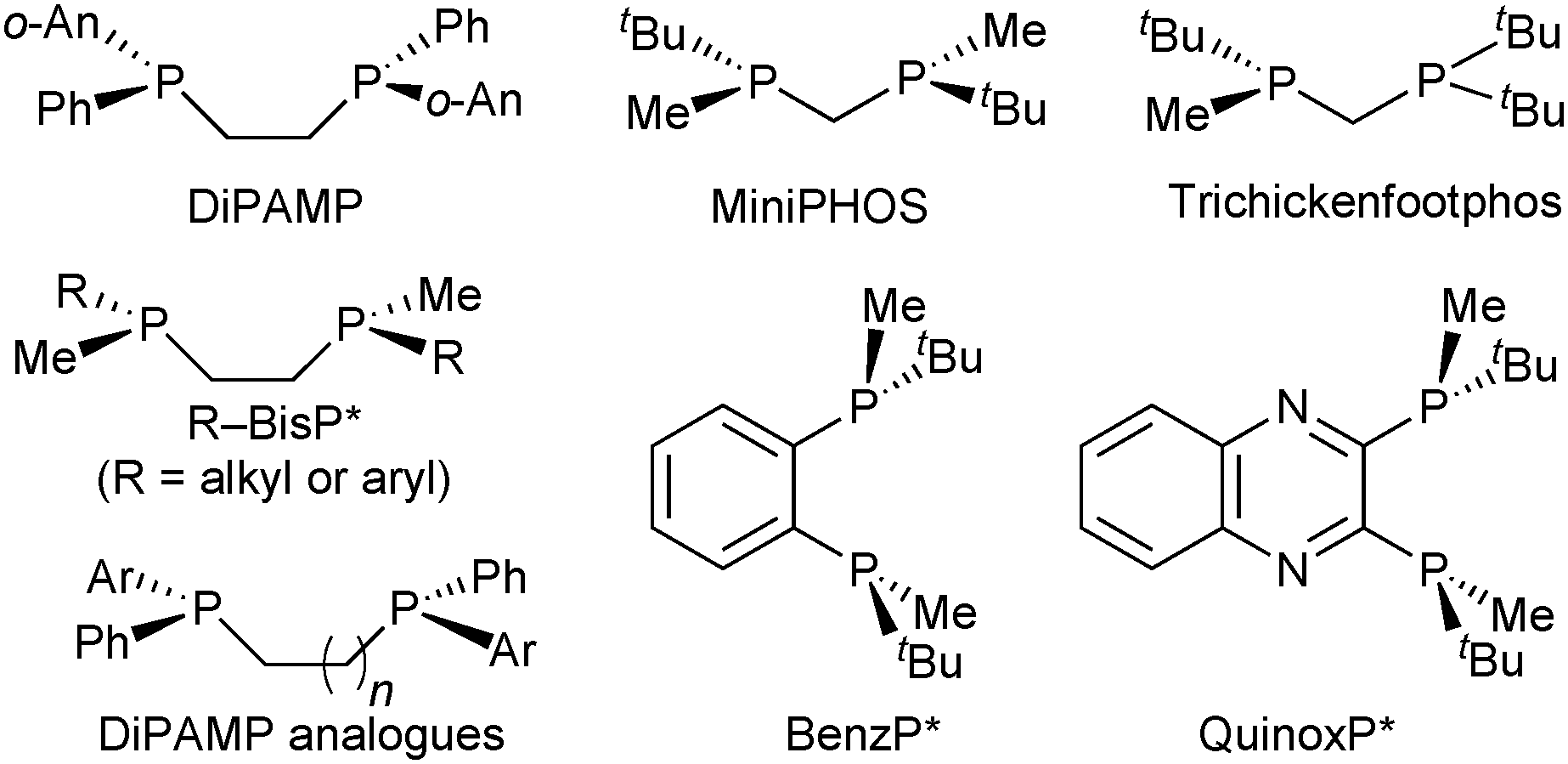 | ||
| Scheme 7 Some frequently used P-stereogenic ligands. | ||
The X-ray structures and DFT calculations of tBu–BisP* complexes exclusively show conformations with the bulky group in the equatorial position,35 but the enantioselectivity of BisP* rhodium catalysts strongly depends on the structure of the substrate.42 Therefore, more rigid backbones were introduced to improve the control of the conformation of the chelate during catalysis. In a first approach, Imamoto used a shorter chelate backbone for ligands of the general formula Me(R)P–CH2–P(Me)R (Scheme 8).43 The four-membered chelate ring formed upon complexation is conformationally rigid and almost perfectly planar.42 MiniPHOS rhodium catalysts outperform BisP* ligands in the hydrogenation of β,β-disubstituted dehydro amino acids (Scheme 8).42
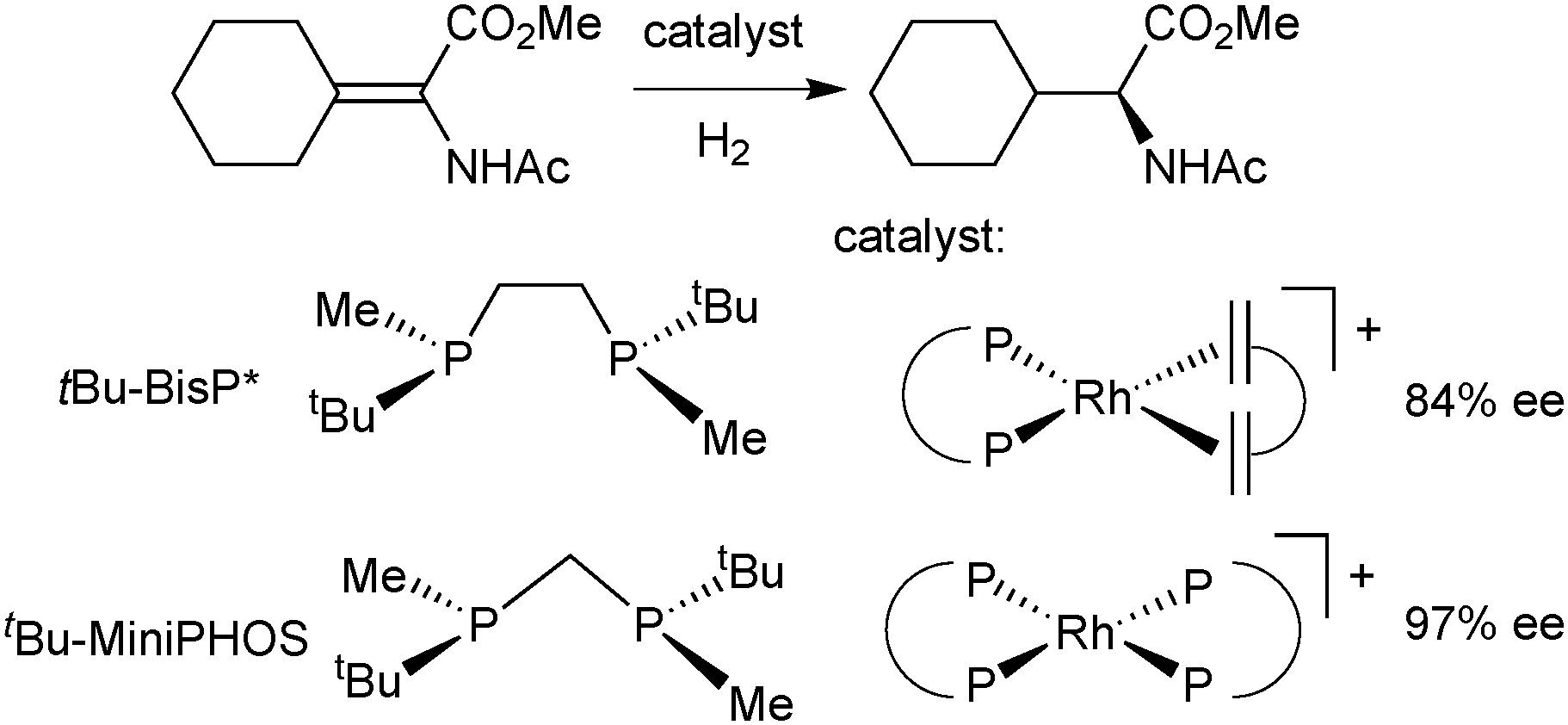 | ||
| Scheme 8 Asymmetric hydrogenation of methyl 2-acetamido-2-cyclohexylideneacetate. | ||
However, MiniPHOS gives lower enantioselectivity than BisP* in the rhodium-catalysed hydrogenation of unsubstituted enamides,44 possibly because its smaller bite angle (ca. 72°) reduces the overall steric bulk. In the C1-symmetric modified MiniPHOS-type ligand tBu2PCH2P(CH3)tBu (Trichickenfootphos, Scheme 7), the large achiral PtBu2 donor apparently compensates for the effect of the small chelate and gives excellent enantioselectivity with dehydro amino acid derivatives.45 The NH-bridged MaxPHOS (RR′P*–NH–P*RR′) is also a MiniPHOS derivative.46
An alternative strategy to rigidify P-stereogenic diphosphines with a five-membered chelate, such as BisP*, is the use of a 1,2-phenylene linker instead of ethane-1,2-diyl. With ligands bearing pseudo-chirality47 on phosphorus such as DuPHOS and BPE, this approach has improved the enantioselectivity of the Rh-catalysed asymmetric hydrogenation of α-acetamidoacrylates (Scheme 9).48
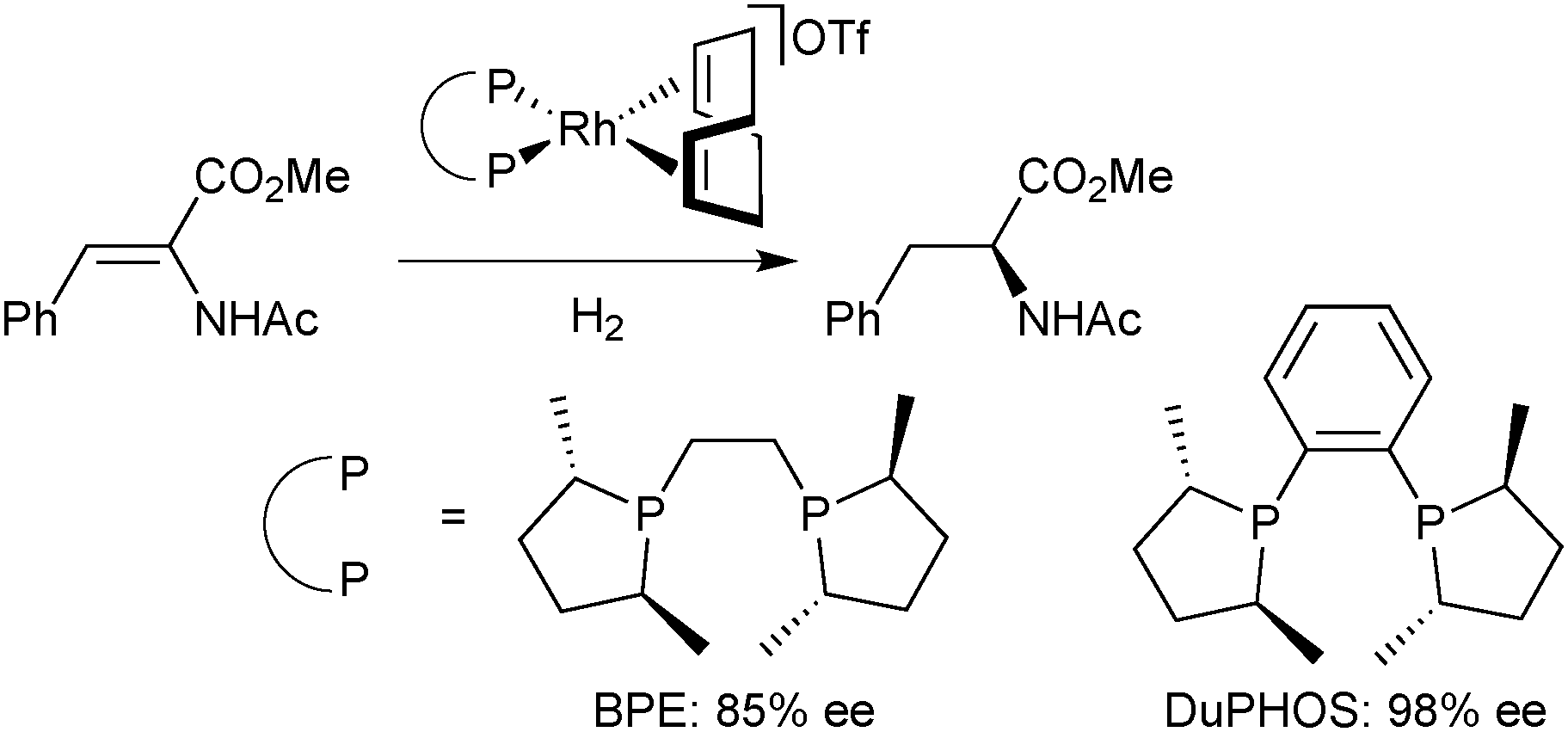 | ||
| Scheme 9 Effect of the linker rigidity on the asymmetric hydrogenation with non-P-stereogenic diphosphines. | ||
On this track, Imamoto prepared P-stereogenic ligands with a 1,2-phenylene backbone such as BenzP*49 and QuinoxP (Scheme 7).50 In the hydrogenation of (Z)-methyl 3-acetamidobut-2-enoate, the enantioselectivity dramatically increased from 19.7% ee with tBu–BisP*41c to 97.6% ee with BenzP* (Scheme 10).49b BenzP* and QuinoxP* have found broad application in asymmetric catalysis, such as Rh-catalysed asymmetric hydrogenation51 and hydroacylation,52 in Cu-catalysed arylation53 and borylation54 reactions, Pd-catalysed allylic alkylations,55 and Fe-catalysed cross coupling.56
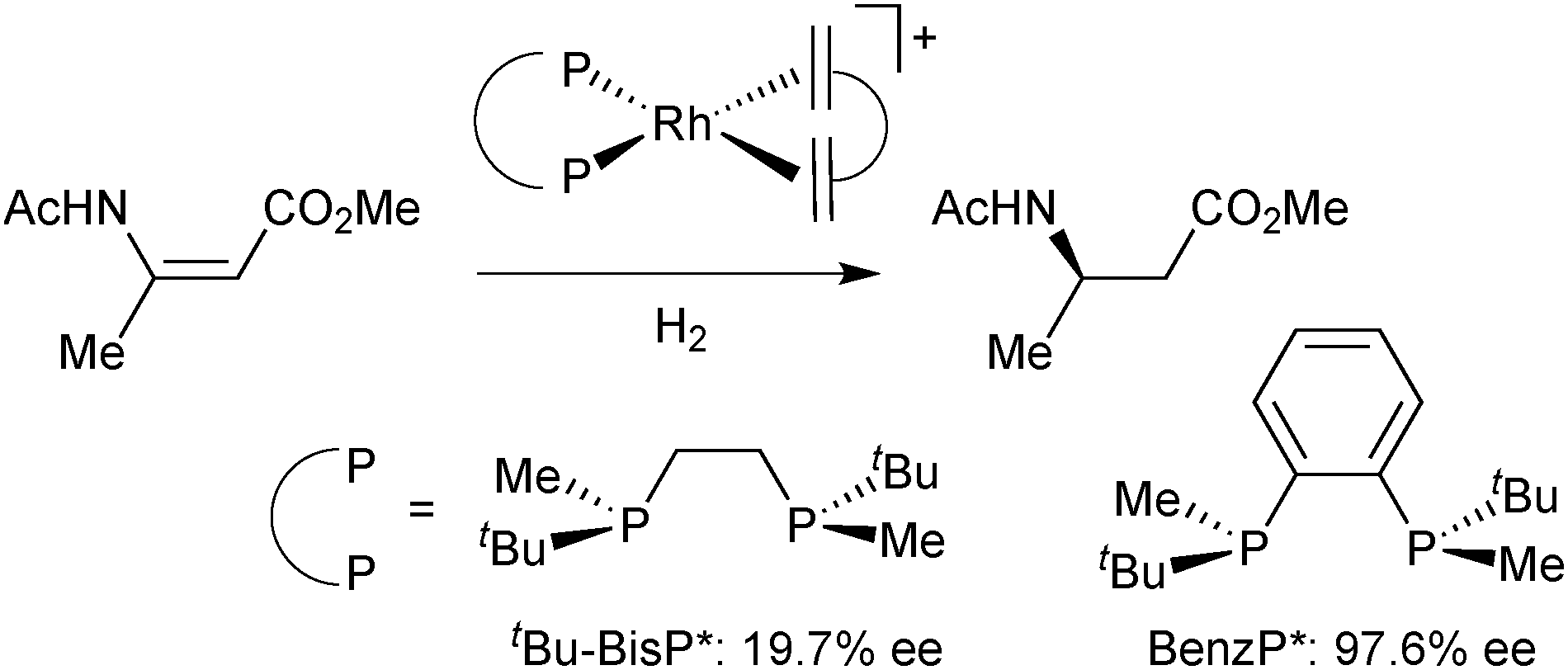 | ||
| Scheme 10 Asymmetric hydrogenation of methyl-(Z)-3-acetamidobut-2-enoate. | ||
Diphosphines with larger chelates
In the context of six-membered phosphacycles, it should be mentioned that caution is required when discussing the conformations of chelate rings formed by multidentate P-stereogenic phosphine ligands, as they lack the strong bias towards the equatorial conformer that is typical of cyclohexanes. This is illustrated by the conformational equilibrium of the phosphinanes (Scheme 11).57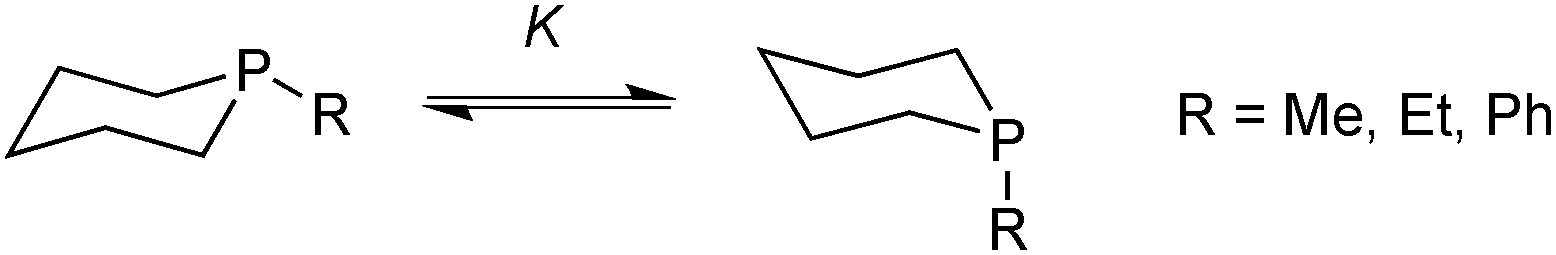 | ||
| Scheme 11 Conformational equilibrium in phosphinanes. | ||
In these molecules, the entropic preference for the axial conformation is larger than in cyclohexanes, and the corresponding ΔH and TΔS values are similar. Hence, the equilibrium constant K for the equatorial/axial equilibrium depends more strongly on temperature. Also, the axial isomer shows weaker 1,3-diaxial interactions because the P–C bonds are longer than C–C. Hence, for 1-methyl, 1-ethyl, and 1-phenyl phosphinanes, the axial conformer is in excess at room temperature.
Back to diphosphines, we have seen above that the most successful P-stereogenic ligands form small chelate rings upon coordination (five- or four-membered, Scheme 8). Six-membered chelate rings have been used with some success in combination with P-stereogenic diarylphosphine donors,58 whereas dialkylphosphine donors have been found to give low enantioselectivity.59 This is unsurprising, as six-membered chelate rings are intrinsically more flexible than smaller ones. The typical example is 2,4-bis(diphenylphosphino)pentane (bdpp), which forms, besides the most stable twist conformation with all-equatorial methyl groups, an energetically accessible achiral half-chair in which the axial substituents on phosphorus assume an achiral conformation (Scheme 12).60
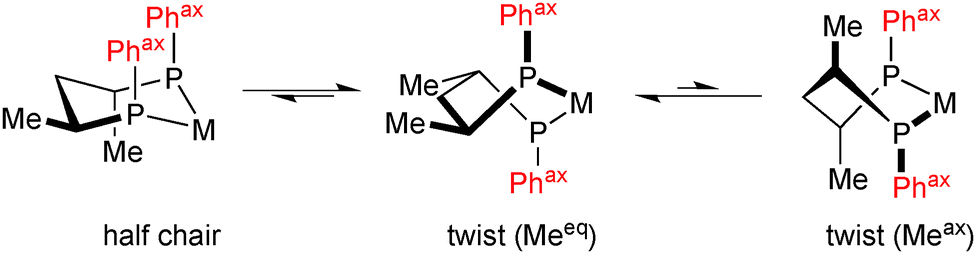 | ||
| Scheme 12 Conformations of (2S,4S)-[RhL2(bdpp)]+ (only one of the two equivalent half chair conformations is shown). | ||
To rigidify P-stereogenic diphosphines with six-membered chelate rings, gem-disubstitution on the central atom of the backbone was attempted with success with diarylphosphine donors, such as in Me2Si(CH2P(1-Np)Ph)2,61 but failed with dialkylphosphines.59 Also, as seen above for five-membered rings, the use of aromatic backbones is beneficial for enantioselectivity. Thus, P-stereogenic diphosphines featuring larger chelates take advantage of rigid xanthene structures.62 With the aim of increasing the bite angle of the P-stereogenic diphosphine, ferrocene-1,1′-diyl also has been used as the backbone, and the resulting ligands give high enantioselectivity in the rhodium-catalysed hydrogenation of acetamido cinnamates and other functionalized alkenes,63 but the rotation of the cyclopentadienyl rings of the ferrocene bridge allows for several conformations of the ligand, as observed by 31P NMR spectroscopy in solution.63b Interestingly, a recent application of a P(tBu)Me/oxazoline ligand in the iridium-catalysed hydrogenation of N-alkyl imines suggests that P-stereogenic phosphines also give stiff conformations in the presence of an external rigidifying element, in this case, a cyclometalated benzimine.64
Overall, the above discussion shows that the conformational issues of P-stereogenic diphosphines play a pivotal role in enantiodiscrimination. Before extending it to tridentate ligands, the synthetic protocols for P-stereogenic building blocks are recapitulated in the next section.
P-Stereogenic synthons
General remarks
The lack of adequate synthetic methods has long been the bottleneck for the application of P-stereogenic ligands in homogeneous catalysis. Therefore, the three established pathways presented in the next section have made a major contribution to alleviate this issue, and the timeline of their development (late 1990s and 2000s) directly precedes the resurgence of catalysts with P-stereogenic ligands. These methods are based on Jugé and Stephan's 1,3,2-oxazaphospholidine boranes derived from ephedrine,65 Evans’66 and Imamoto's67 enantioselective deprotonation of phosphine boranes with sparteine, and Mislow and Han's menthyl phosphinates.68 For an exhaustive review of all known synthetic methods, the reader is referred to dedicated surveys.11,3,2-Oxazaphospholidine boranes
In Jugé's method, enantiomerically pure phosphines are prepared from an aryl diaminophosphine and ephedrine via a 1,3,2-oxazaphospholidine borane.65 Ring-opening of oxazaphospholidine boranes with a wide scope of organolithium reagents65c gives the corresponding aminophosphine boranes (Scheme 13).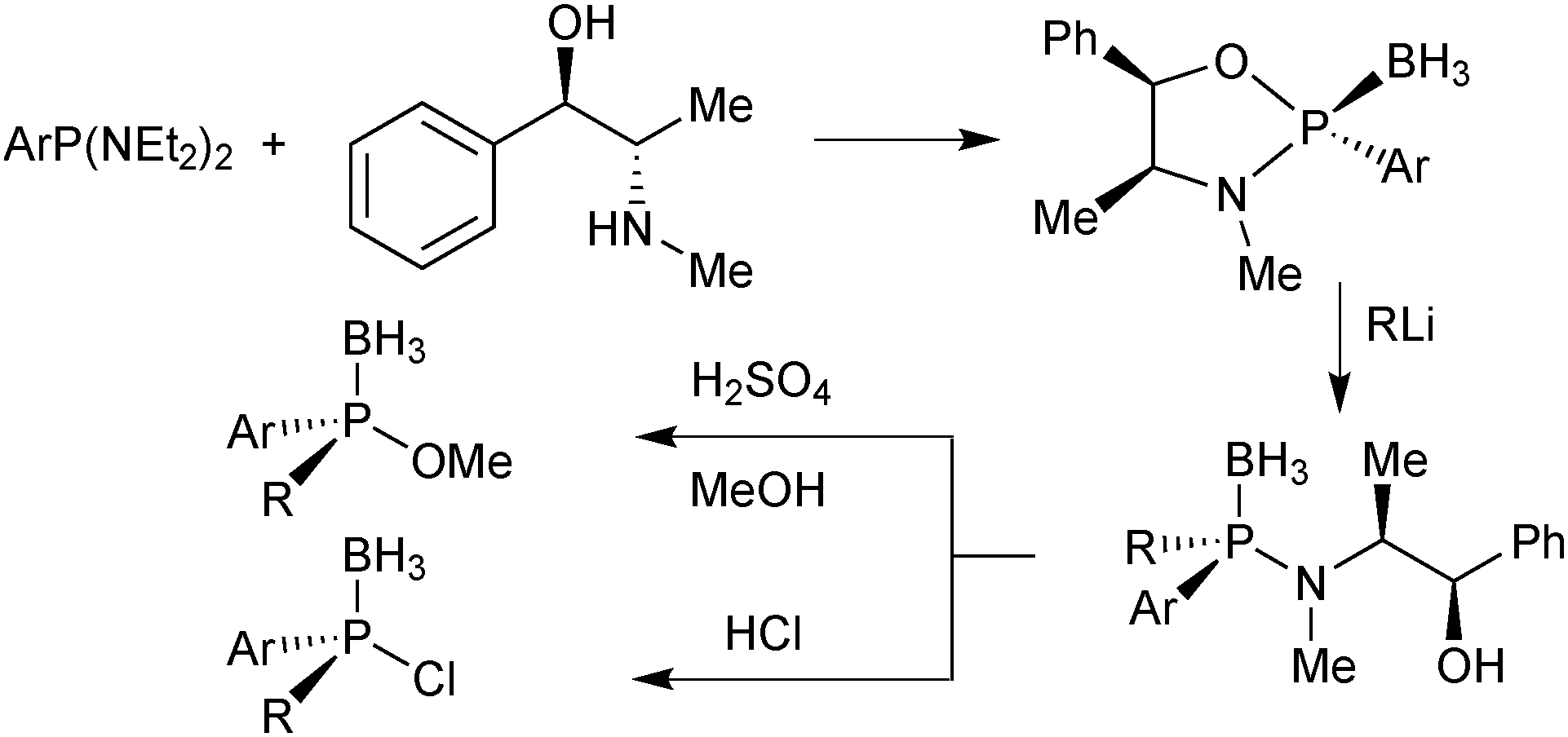 | ||
| Scheme 13 Jugé's enantiomerically pure aminophosphine and phosphinite boranes. | ||
Also bulky alkyl lithium reagents can be used,39 but supermesityllithium (2,4,6-tri-tert-butylphenyl lithium) or 2-adamantyllithium fails to give the desired product.69 Organomagnesium compounds react sluggishly and require heating, which decreases the enantiospecificity.65b Usually, the aminophosphine boranes are obtained as single diastereoisomers after a simple recrystallization step. The aminophosphine is then methanolised under acidic conditions into a methyl phosphinite borane.65 Alternatively, it can be treated with anhydrous HCl in an aprotic solvent such as toluene to give the more reactive chlorophosphine borane.70 Both compound classes react as electrophiles, and after an SN2@P reaction with an organolithium reagent, tertiary phosphine boranes are obtained (Scheme 14). Alkyl- as well as aryllithium reagents react with high enantiospecificity.65
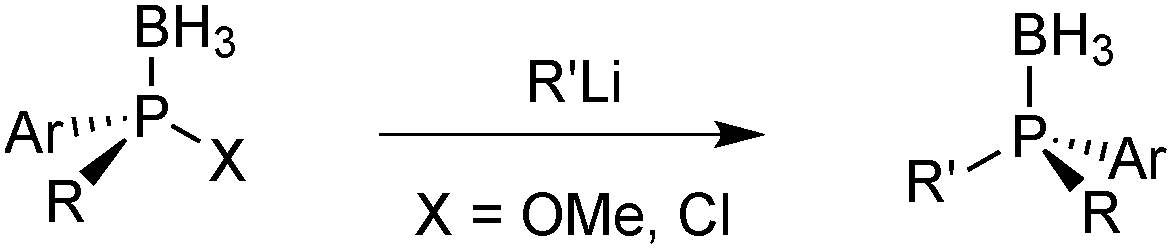 | ||
| Scheme 14 Nucleophilic substitution on methyl phosphinite boranes and chlorophosphine boranes. | ||
One advantage of the electrophilic chlorophosphine boranes is that they undergo umpolung to nucleophilic secondary lithium phosphide boranes feasible by transmetalation with tBuLi at −85 °C (Scheme 15).65c
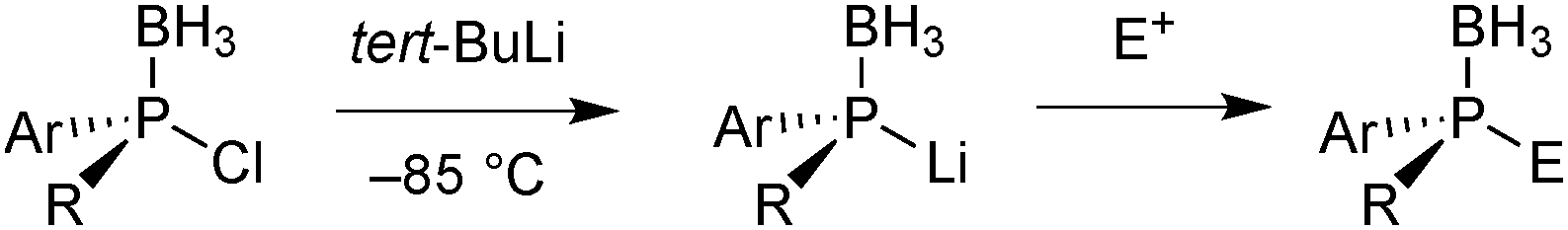 | ||
| Scheme 15 Umpolung of the chlorophosphine borane with tBuLi and quenching with an electrophile. | ||
Deboronation of the final product is achieved either by reaction with an excess of amine71 or by treatment with strong acids such as HBF4 etherate.72 The latter method is mandatory for trialkylphosphines due to their electron-rich nature. The Jugé–Stephan method is generally applicable when at least one of the substituents in the target (tertiary) phosphine is phenyl. A drawback of the method is the use of ephedrine, which is nowadays a restricted chemical due to its use in illicit drug manufacturing.
Enantioselective deprotonation with sparteine
Sparteine-mediated enantioselective deprotonation has been developed by Evans and Imamoto. In 1995, Evans demonstrated that the reaction of an aryldimethylphosphine borane with sBuLi/(−)-sparteine leads to the enantioselective deprotonation of a methyl group, which was then exploited to prepare diphosphine ligands (Scheme 16).66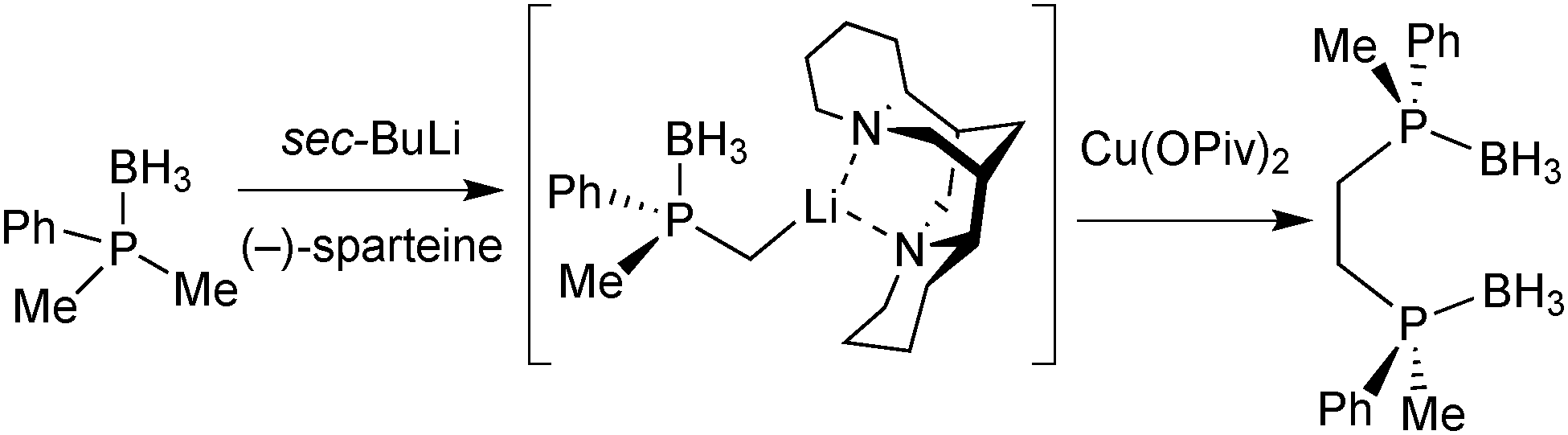 | ||
| Scheme 16 Evans’ enantioselective deprotonation with (−)-sparteine. | ||
Imamoto exploited Evans’ enantioselective deprotonation to prepare enantiomerically pure secondary phosphine boranes.67 To this goal, the intermediate organolithium derivative was oxidised with O2 to the hydroxymethylphosphine borane with retention of configuration. Ruthenium-catalysed oxidation to the carboxylic acid and spontaneous decarboxylation under basic reaction conditions give secondary phosphine boranes with formal inversion (Scheme 17).
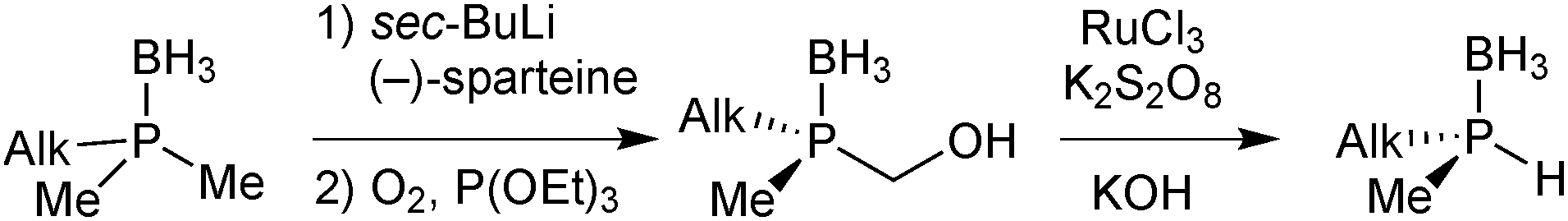 | ||
| Scheme 17 Imamoto's preparation of secondary phosphine boranes. | ||
The secondary phosphine boranes are stable towards air and moisture, and are valuable precursors for the synthesis of a broad variety of ligands as described below.73 A drawback is the stoichiometric amount of sparteine, whose (−)-enantiomer has experienced a global shortage since the early 2010s.74 Nowadays, both enantiomers of the alkaloid are commercially available, at prices typical for fine chemicals.
Menthyl phosphinates
Early examples of optically active tertiary phosphines relied on resolution of the corresponding racemic oxides or quaternary phosphonium salts and subsequent reduction,75 which restricts the scope of available enantiomerically pure phosphines. In 1967, Mislow tackled the problem by using menthol as a chiral auxiliary: the diastereoisomers of its phosphinate esters are easily resolved.76 Substitution of the menthyl group with alkyl or aryl Grignards then gives the tertiary phosphine oxides with high stereospecificity (Scheme 18). This method enabled Knowles to prepare the DiPAMP ligand, which proved to be so successful in the Rh-catalysed enantioselective hydrogenation of olefins (see above).33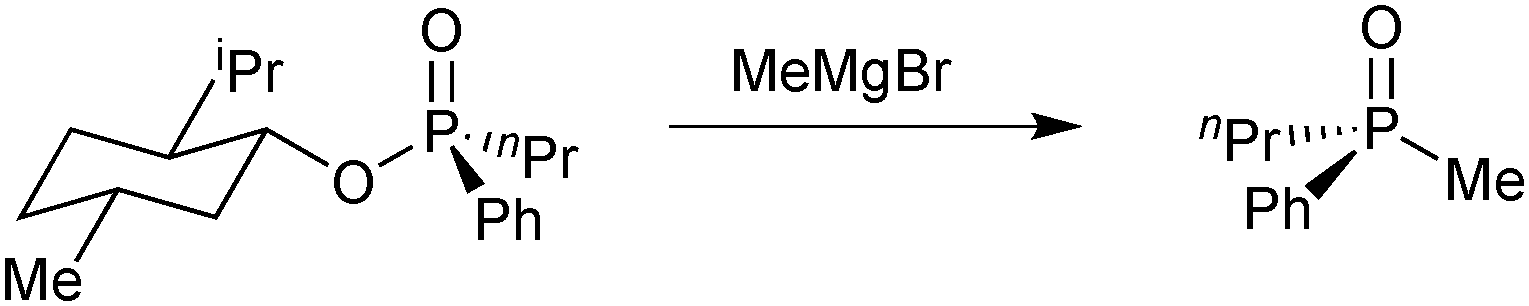 | ||
| Scheme 18 Mislow's synthesis of enantiomerically pure methylphenyl-n-propylphosphine oxide. | ||
However, efforts directed towards preparing enantiomerically pure secondary phosphine oxides – which, after deprotonation, offer a versatile synthon that reacts with a broad range of electrophiles – remained limited,77 possibly due to the belief that the deprotonated secondary phosphine oxide anion is inherently stereolabile.78
This prejudice was overturned by Han. In 2008, he reported that the rapid epimerization of (−)-menthyl (RP)-phenylphosphinate and its substitution products under alkaline conditions is not due to the stereolability of the secondary phosphine oxide anion, but originates from the reaction of the phosphinate ester with a metal alkoxide or hydroxide.68 Thus, by carefully ensuring that no hydroxide or alkoxide is formed under the reaction conditions (e.g. with a menthol-free starting material), he prepared a variety of secondary and tertiary phosphine oxides with excellent enantiospecificity (Scheme 19).
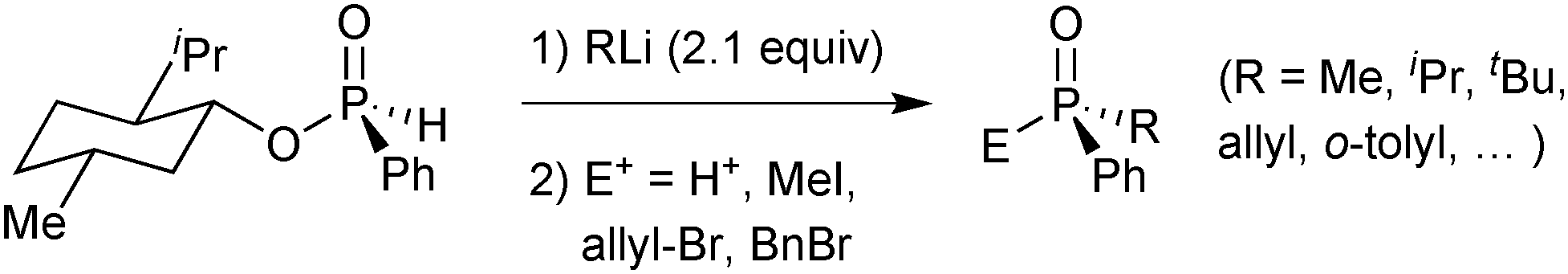 | ||
| Scheme 19 Han's preparation of secondary and tertiary phosphine oxides from menthyl H-phosphinates. | ||
The method has been applied, for example, in the preparation of organocatalysts for the Morita–Baylis–Hillman reaction79 and of P-stereogenic macrocyclic N2P2 ligands.80 It is most successful when one of the substituents in the target (tertiary) phosphine is an aryl group, and is thus often an alternative to the Jugé–Stephan method. Also, menthol is cheap and readily available, and the reaction is easily scaled up. One drawback is the use of a phosphine oxide as the protecting group, though. Although a wealth of literature exists on the (stereospecific) reduction of secondary and tertiary phosphine oxides,81 the reduction conditions need to be tuned to the specific system and are less general than for the deprotection of phosphine boranes.
P* pincer catalysts of metals other than iron
PNP catalysts
The first PNP pincer ligands based on the pyridine backbone were prepared nearly 20 years ago (Scheme 21), and have found little attention since then. Zhang used Jugé's protocol65 for his P(o-An)Ph-based ligand, which gave moderate enantioselectivity in the palladium-catalysed allylic alkylation and ruthenium-catalysed hydrosilylation.82 Inspired by Evans’ enantioselective deprotonation,66 Livinghouse developed a dynamic resolution/alkylation of the secondary tert-butylphenylphosphine borane to prepare the corresponding PNP pincer (Scheme 20).83 Castillon applied this ligand in the ruthenium-catalysed hydrogenation of ketones with moderate to good enantioselectivity (60–95% ee at −40 °C).84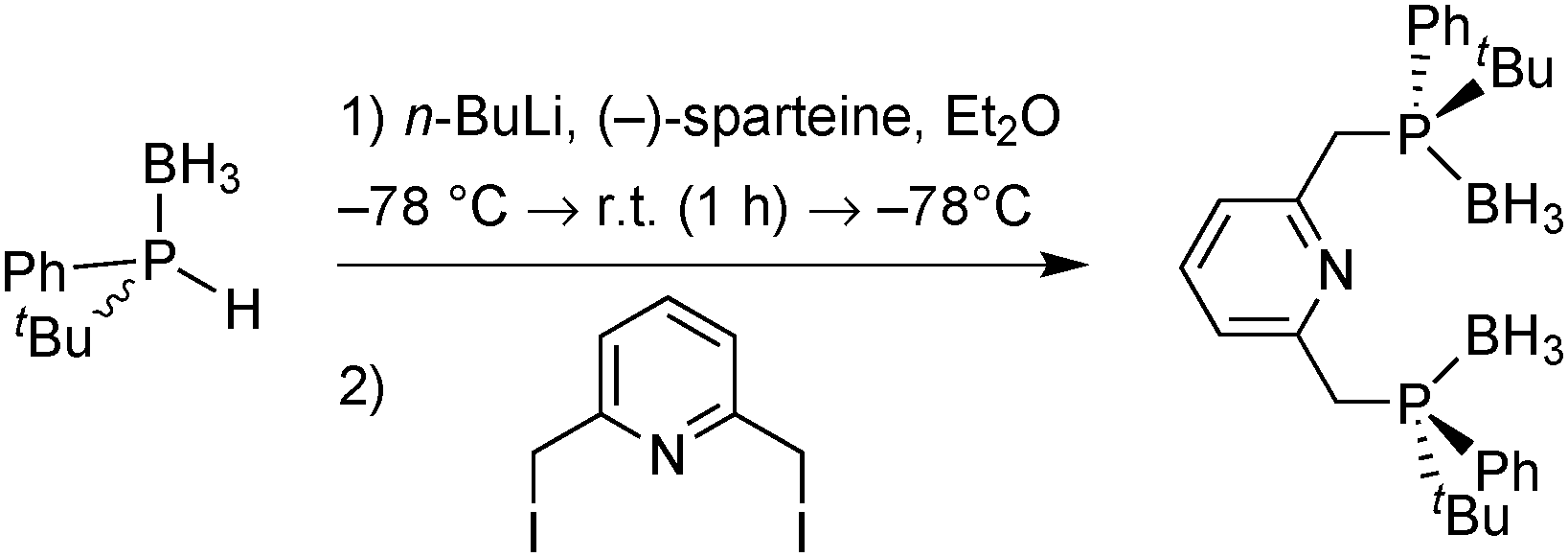 | ||
| Scheme 20 Livinghouse's P* pincer synthesis by dynamic resolution/bisalkylation. | ||
Imamoto prepared the P(tBu)Me analogue and used it in a variety of enantioselective reactions.85 These encompass Pd-catalysed Michael additions (1 out of 19 substrates gave >90% ee)85b and intramolecular hydroamination (up to 47% ee),85e Ir-promoted ketone hydrogenation (17% ee),85c and aza-Michael addition with a Ni catalyst (up to 46% ee).85d
PCP catalysts
The first P-stereogenic PCP ligand, 1,3-bis(tert-butylphenylphosphinomethyl)benzene,86 prepared in 2001 by van Koten with the dynamic resolution/alkylation protocol,83 gave 18% ee in the Ru-catalysed ATH of acetophenone.86c More recently, Imamoto used enantiomerically pure tert-butylmethylphosphine borane to obtain the P(tBu)Me analogue, which gave moderate enantioselectivity (up to 83% ee) in the Pd-catalysed addition of diarylphosphines to nitroalkenes.87 The application in asymmetric catalysis of these PNP and PCP pincer ligands is discussed below in the context of our results with iron(II).P* pincer catalysts of iron(II)
Ligand scope and synthons
For our studies, we chose either the 2,6-dimethylpyridine (PNP, 1)88,89 or diethylamine backbones (PN(H)P, 2)90 in combination with P(Cy)Me (a), P(tBu)Me (b), and P(Ph)Me (c) as P-stereogenic donors (Scheme 21).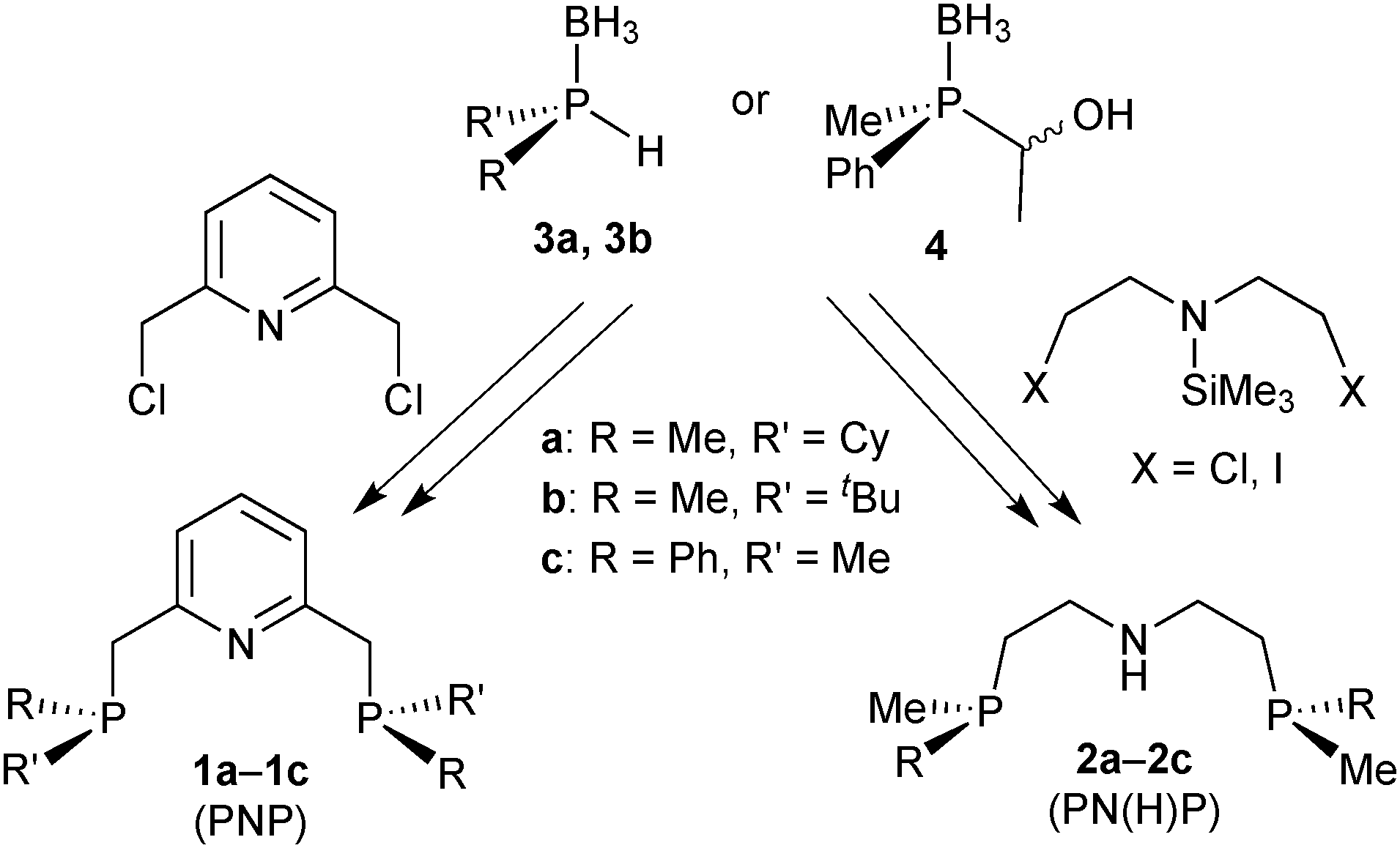 | ||
| Scheme 21 General synthetic pathway to the P-stereogenic pincers. | ||
Enantiomerically pure tert-butylmethylphosphine borane (3b) was prepared by Imamoto's procedure (Scheme 17). As the iron(II) complexes of the P(tBu)Me based pincer 1b turned out to be unstable (see below), we also targeted its P(Cy)Me analogue 3a. Imamoto's method gives (R)-cyclohexyl(hydroxymethyl)methylphosphine borane (5a) with 75% ee only, though.67 Conversion of 5a to the crystalline p-phenylbenzoyl ester and crystallisation gave 7a as a single enantiomer. Thus, enantiomerically pure cyclohexylmethylphosphine borane (R)-3a was obtained after saponification and Ru-catalysed oxidation (Scheme 22).88
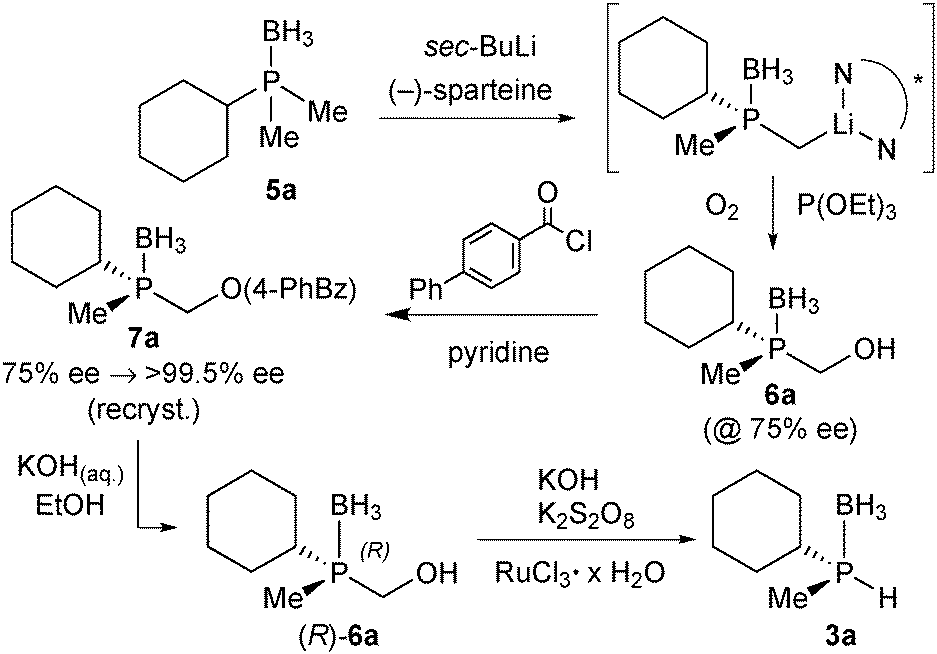 | ||
| Scheme 22 Synthesis of enantiomerically pure (S)-cyclohexylmethylphosphine borane 7a. | ||
Finally, DFT calculations (see below) prompted us to prepare the P(Me)Ph-based pincers 1c and 2c. In this case, Imamoto's approach in Scheme 17 is not suitable, as methylphenylphosphine borane cannot be prepared as in Scheme 23 because the intermediate of type 6 undergoes overoxidation to the corresponding phosphinite borane P(Me)(Ph)(OH)·BH3.67 Also, methylphenylphosphide borane has been found to be configurationally unstable.91 Therefore, we developed an alternative approach inspired by Buono's masked secondary phosphine boranes (4);92 we treated diastereomerically pure (RP)-(−)-menthyl-H-phenylphosphinate (prepared on a multi-gram scale from (−)-menthol and PhPCl2)93 with MeLi, quenched the lithium salt of the methylphenylphosphine oxide with acetaldehyde, and reduced (SP)-(1-hydroxyethyl)methylphenylphosphine oxide 8 with BH3·SMe2 to (SP)-(1-hydroxyethyl)methylphenylphosphine borane (4) with formal retention (Scheme 23).94 This route is easily scalable, and (SP)-4 was routinely prepared on a >5 g scale.89
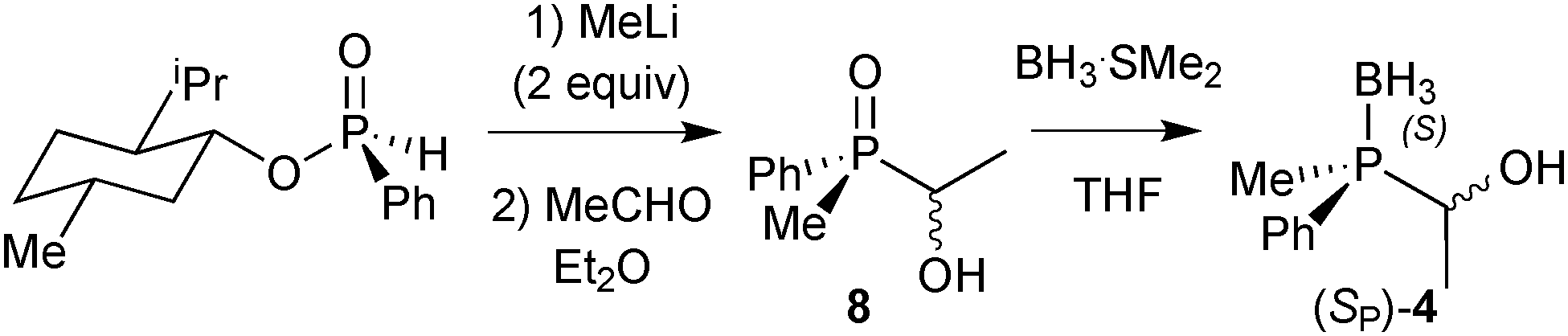 | ||
| Scheme 23 Preparation of masked secondary phosphine borane (SP)-4 from (RP)-menthyl phenylphosphinate. | ||
Pyridine based pincer complexes
PNP ligand synthesis
The P(Me)R·BH3 (R = Cy, 3a: R = tBu, 3b) units were installed onto the 2,6-dimethylpyridine backbone to give the borane-protected PNP pincer ligands 9a and 9b following Imamoto's procedure (Scheme 24).85a The phenylmethylphosphine pincer 9c was prepared by base induced deacylation of 4, followed by double alkylation with 2,6-bis(chloromethyl)pyridine.89 Ligand 9c has been previously prepared by a different procedure on a small scale (<50 mg) as a 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :42 C2:meso diastereomeric mixture (84% ee for the C2 diastereomer).95
:42 C2:meso diastereomeric mixture (84% ee for the C2 diastereomer).95
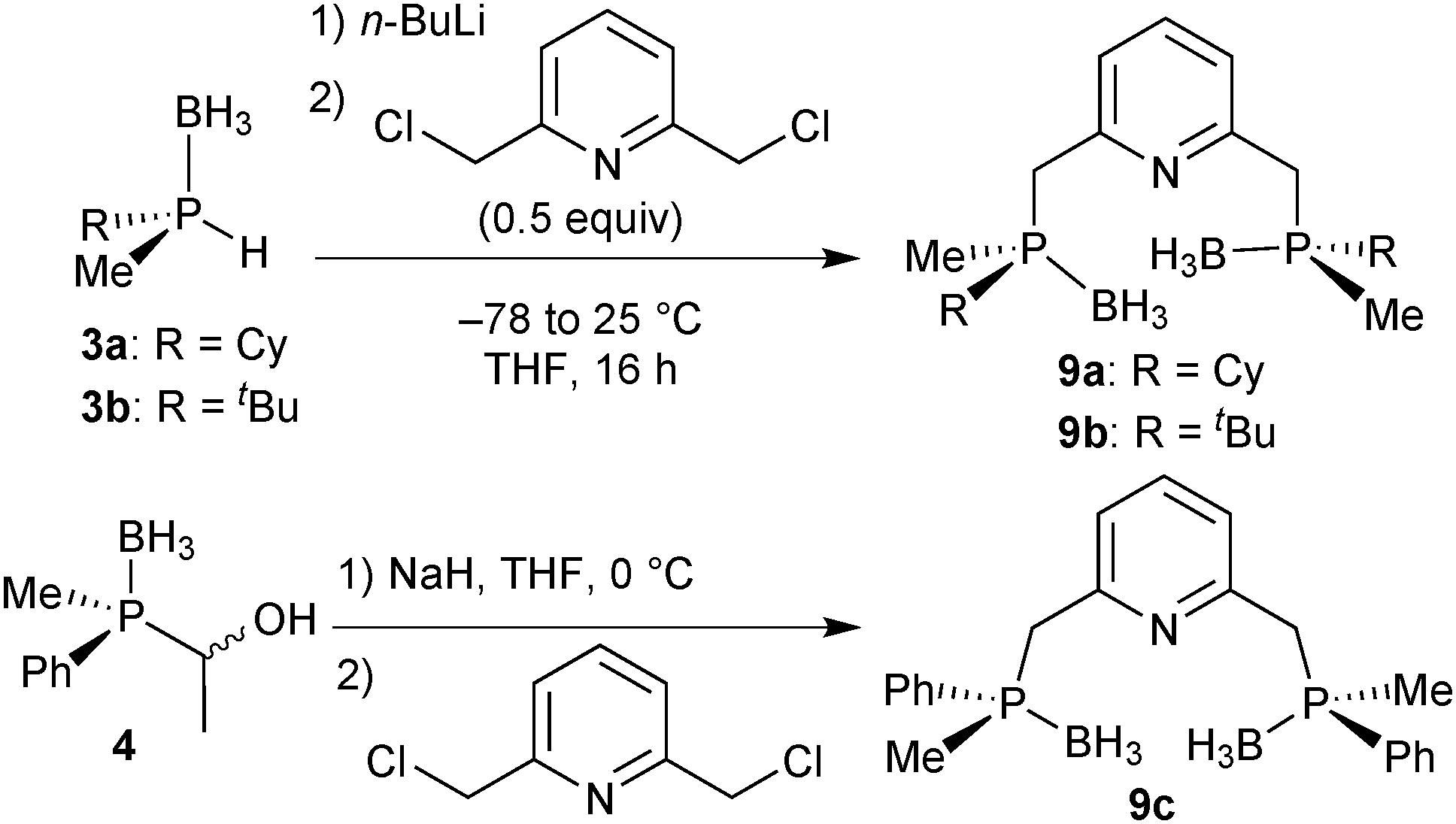 | ||
| Scheme 24 Preparation of borane-protected PNP pincer ligands 9a–9c. | ||
[FeBr2(CO)(PNP)]
The borane adducts 9a–9c were deprotected with HBF4·OEt2 in dichloromethane72 to the free pincer ligands 1a–1c, which were treated with a suitable iron source (FeBr2 or [FeBr2(PPh3)2]) under a CO atmosphere to give the iron dibromocarbonyl complexes mer,trans-[FeBr2(CO)(1a–1c)] (10a–10c) (Scheme 25).88 These complexes are typically deep blue, whereas the cis dibromo analogues are red (see below). Complexes 10a–10c are stable towards air and moisture. In the work-up, 10a is washed with water in air. However, the stability of 10a–10c decreases with increasing steric bulk of the substituents on phosphorus. Thus, the tert-butylmethylphosphine derived complex 2b slowly decomposes both in solution and in the solid state.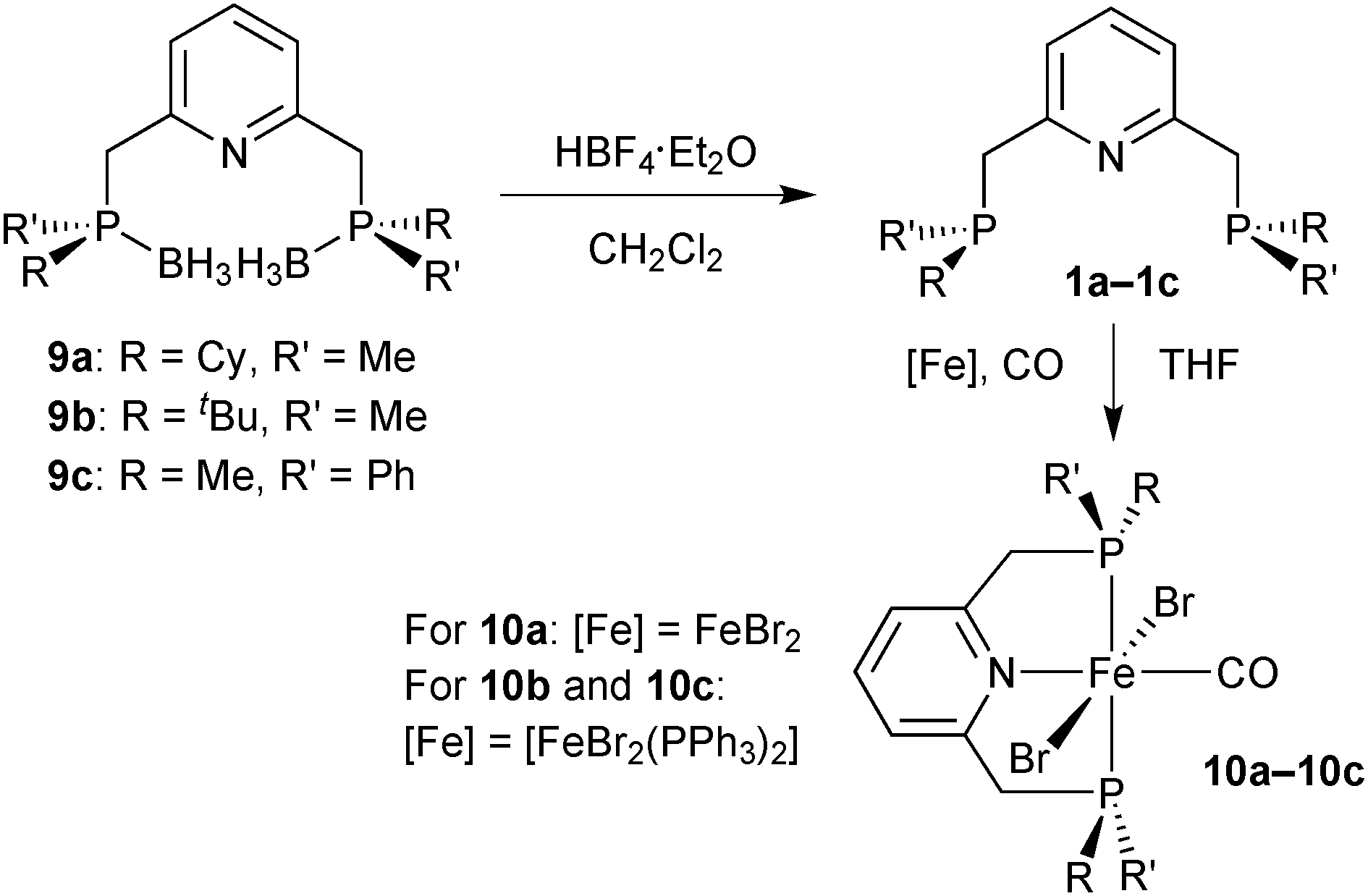 | ||
| Scheme 25 Preparation of iron dibromocarbonyl complexes 2a–2c. | ||
The X-ray structure of 10a (Fig. 1) reveals that the pyridine-pincer backbone of 1a is tilted to enable the envelope conformation for the two chelate rings.88
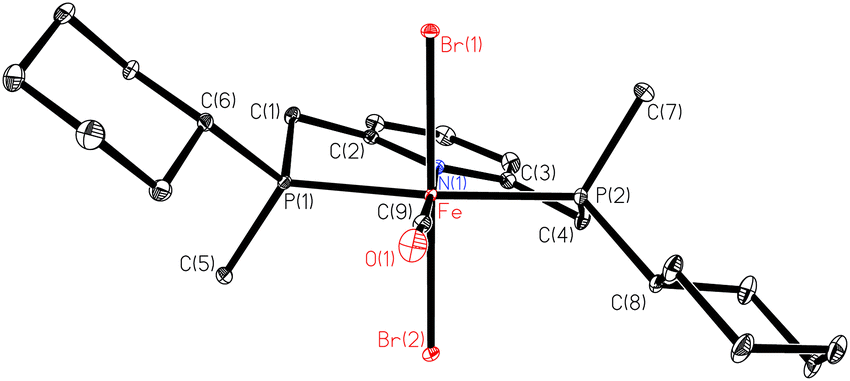 | ||
| Fig. 1 ORTEP drawing of 10a (reprinted with permission from ref. 88. Copyright 2018 American Chemical Society). | ||
Depending on the stereocentre at phosphorus, the tilt is either λ (as for 10a with (S,S)-1a) or δ (observed for 10c with (R,R)-1c), and the large substituent on phosphorus occupies a pseudo-equatorial position.
Hydride complexes
The dibromocarbonyl complexes 10a–10c react with NaBHEt3 (1 equiv.) in THF or CH2Cl2 to give the bromohydridocarbonyl complexes [FeBrH(CO)(PNP)] (11a–11c) (Scheme 26). In the major isomer of 11a–11c, the hydride ligand is located trans to bromide.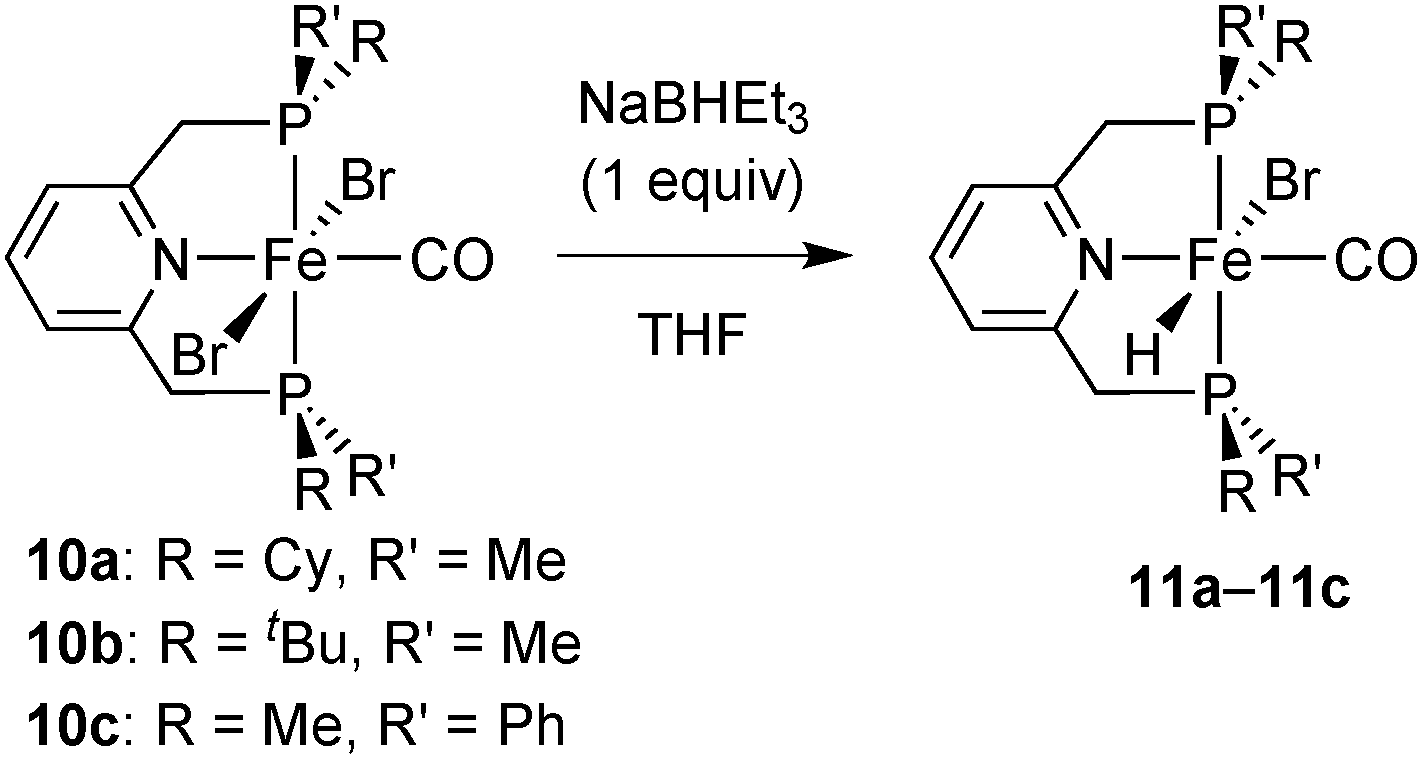 | ||
| Scheme 26 Synthesis of hydrides 11a–11c. | ||
All hydrides are extremely sensitive towards air and moisture, and the P(tBu)Me derivative 11b was not isolable in reasonable purity. As hydrides 11b–11c decompose in solution, they were always freshly prepared for catalytic experiments. A putative decomposition product (among unidentified brown precipitates) is an iron(0) dicarbonyl complex, as observed for Fe complexes of the achiral 2,6-bis(diisopropylphosphinomethyl)pyridine pincer.16 As for dibromocarbonyl complexes, bulkier phosphines (e.g. tert-butyl substituted ones) form less stable complexes.
Due to its potential role in catalysis (see below), we also prepared dihydride 12a (Scheme 27). The major species contains trans hydrides, but the other components of the reaction mixture were not characterised. The dihydride is highly sensitive towards air and moisture and decomposes in solution.
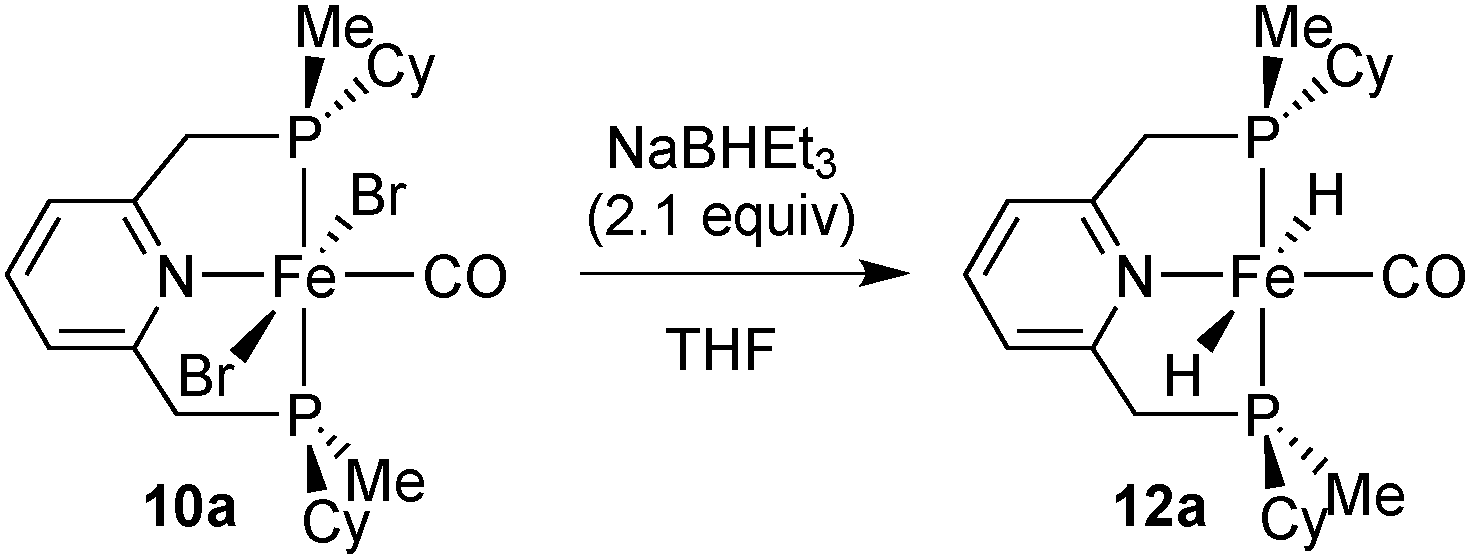 | ||
| Scheme 27 Preparation of iron dihydridocarbonyl complex 12a. | ||
Addition of a stoichiometric amount of acetophenone to 12a did not change the major species present in a C6D6 solution as observed by NMR spectroscopy, and only trace amounts (<2%) of phenylethanol were formed (which might originate from impurities of 12a). A similar observation has been made for an achiral Fe/PNP dihydride.96 This indicates that the dihydride 12a is not involved in the catalytic cycle of ketone hydrogenation with such complexes.
Hydrides 11a–11c in asymmetric hydrogenation
After dehydrobromination with base (KOtBu), the hydride complexes 11a–11c catalyse the hydrogenation of ketones under H2 pressure.88 The best results obtained for acetophenone (13) as a model substrate are given in Scheme 28. Under optimised conditions, catalyst 11a achieved full conversion of 13 to (S)-1-phenylethanol (14). The tert-butylmethylphosphine derived complex 11b and phenylmethylphosphine derived 11c gave lower conversion (70 and 28%, respectively). Overall, the enantioselectivity was modest and reached 48% ee with the cyclohexylmethylphosphine derived pincer 11a.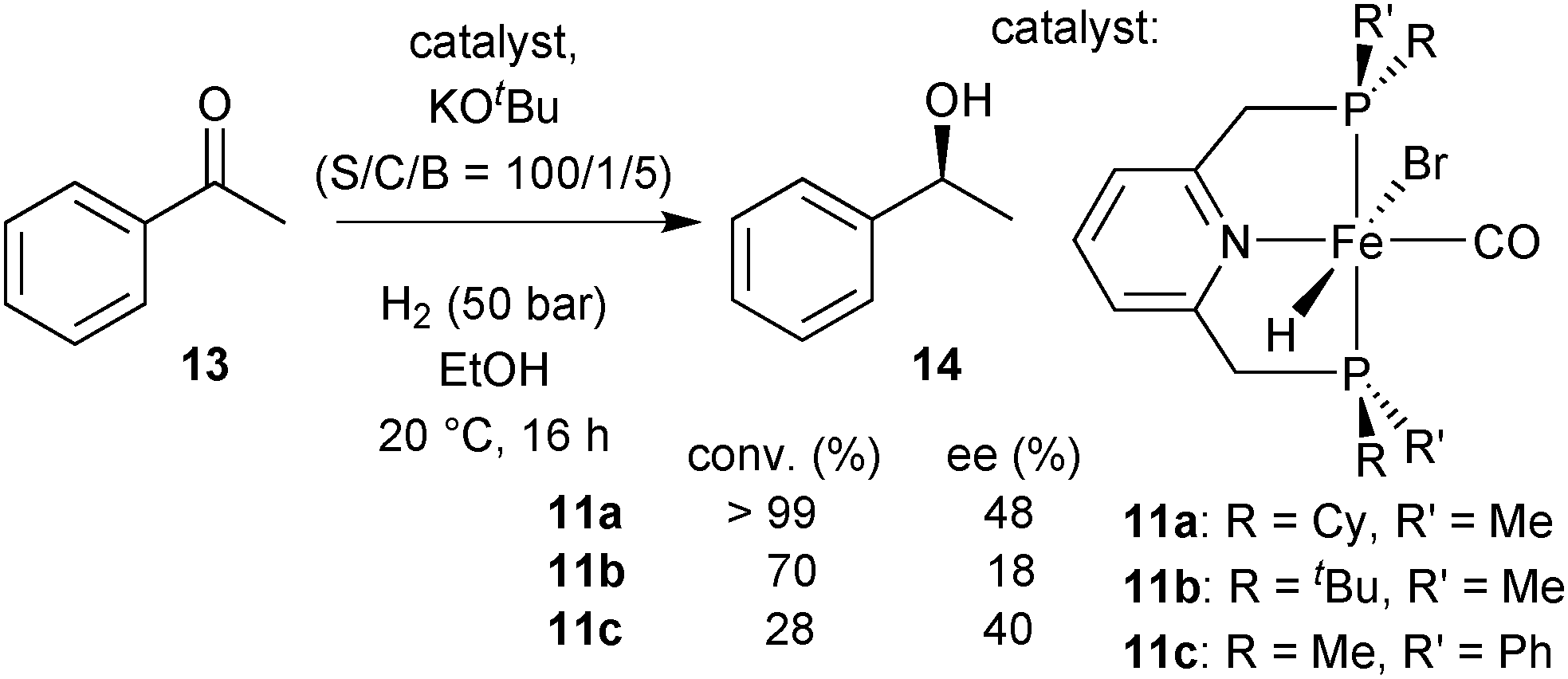 | ||
| Scheme 28 Acetophenone hydrogenation with 11a–11c as catalysts. | ||
DFT study on [FeHBr(CO)(1a)], 11a
To improve the ligand design, we were confronted with the need for a stereochemical model for enantiodiscrimination. This involved us in a long-standing mechanistic dispute concerning the related achiral P(tBu)2 based iron(II) catalyst for ketone hydrogenation,16 for which different mechanisms have been studied (Scheme 29). Milstein's original suggestion of an inner sphere mechanism (I)16 has been challenged by Yang and Hopmann, who proposed outer sphere mechanisms of type D based on a dihydride complex (12).96 | ||
| Scheme 29 Proposed mechanisms for the hydrogenation of ketones with 11a. | ||
Eventually, Milstein put forward the outer-sphere mechanism O, which involves the iron(0) species 15 whose chiral analogue is shown in Scheme 30.97 The base dehydrobrominates precatalyst 11a to give a dearomatised intermediate that reductively eliminates H+. Rearomatisation and EtOH coordination give the iron(0) species 15, which then transfers a benzylic H atom from the PNP ligand as hydride to the carbonyl group of acetophenone (Scheme 30, D). A hydrogen bond involving the coordinated ethanol molecule directs the incoming substrate and activates its carbonyl group towards nucleophilic attack, which is reminiscent of the well-established bifunctional mechanism.23
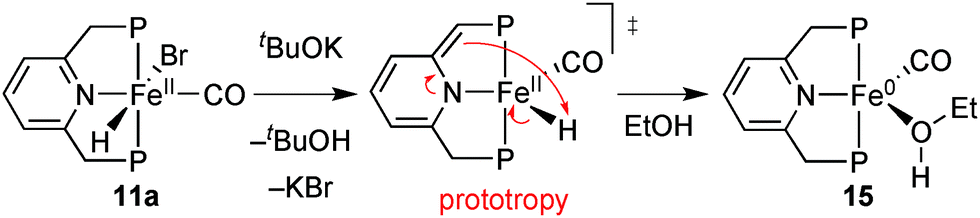 | ||
| Scheme 30 Formation of the iron(0) species 15 from 11a. | ||
We reasoned that the additional stereochemical information provided by the chiral ligand may help discriminate between these mechanisms, and hence undertook a comprehensive DFT study by modelling the enantiodetermining steps of the three mechanisms in Scheme 30.88 Notably, only the outer-sphere mechanism D predicts the experimental sense of induction and the enantioselectivity, in agreement with Milstein's proposal which is mostly based on kinetic considerations.97 Beyond contributing to assess the mechanism, the DFT study revealed the conformational issues discussed below.
Pincer ligand conformation
The X-ray structure of dibromocarbonyl 10a shows that the 5-membered rings P(1)–Fe–N(1)–C(2)–C(1) and P(2)–Fe–N(1)–C(3)–C(4) assume envelope conformations with the P atoms in the endo position and equatorial cyclohexyl groups (Fig. 1). The torsion angles P(1)–Fe–N(1)–C(2) (θ1) and P(2)–Fe–N(1)–C(3) (θ2) describe the conformation of the five-membered chelate rings in the iron-bound PNP pincer (Fig. 2).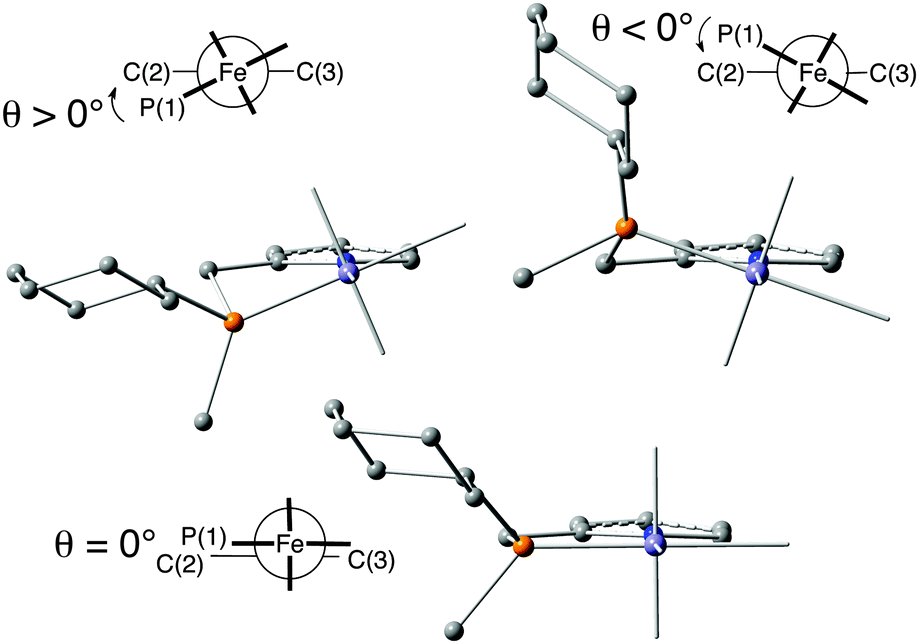 | ||
| Fig. 2 Conformation of the five-membered chelate ring (with Newman projection and dihedral angle θ) (reprinted with permission from ref. 88. Copyright 2018 American Chemical Society). | ||
The conformation of the chelate ring determines the position of the P-substituents (equatorial or axial in the envelope conformation or inclinal in the planar one)32b and the tilt of the pyridine ring with respect to the Fe/PNP plane. The inversion of the ring swaps the cyclohexyl and methyl P substituents between equatorial and axial positions and tilts the pyridine ring with respect to the P(1)–Fe–N(1) and P(2)–Fe–N(1) planes. Positive torsion angles (θ1 and θ2) correspond to equatorial cyclohexyl groups and to the λ conformation of the backbone. For negative θ values (−12° to −27°), the cyclohexyl is axial, and the backbone is in the δ conformation. The calculations indicate that the five-membered chelate ring can also assume a planar conformation (θ close to 0°) with inclinal substituents.
In the enantiodetermining step of Milstein's unprecedented outer-sphere mechanism O, a benzylic H atom of the ligand is transferred as hydride from the five-coordinate iron(0) complex [Fe(CO)(EtOH)(1a)] (15) to acetophenone (Scheme 30O). Complex 15 exists in two different conformations of the chelate ring (15a and 15b). The approaching acetophenone is directed by the incipient C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Fe interaction and by the CO⋯HOEt hydrogen bond to the ethanol ligand. As acetophenone approaches the iron(0) complex 15a/15b, the conformation of the chelate ring changes to allow the attack of the benzylic H atom as hydride onto the carbonyl C atom (Fig. 3). The conformation determines from which benzylic position the hydride is transferred, and acetophenone approaches with either enantioface, for a total of four transition states, O(1)–O(4). Intriguingly, the most stable TS O(1) features axial cyclohexyl groups and hence the less stable conformation.
O⋯Fe interaction and by the CO⋯HOEt hydrogen bond to the ethanol ligand. As acetophenone approaches the iron(0) complex 15a/15b, the conformation of the chelate ring changes to allow the attack of the benzylic H atom as hydride onto the carbonyl C atom (Fig. 3). The conformation determines from which benzylic position the hydride is transferred, and acetophenone approaches with either enantioface, for a total of four transition states, O(1)–O(4). Intriguingly, the most stable TS O(1) features axial cyclohexyl groups and hence the less stable conformation.
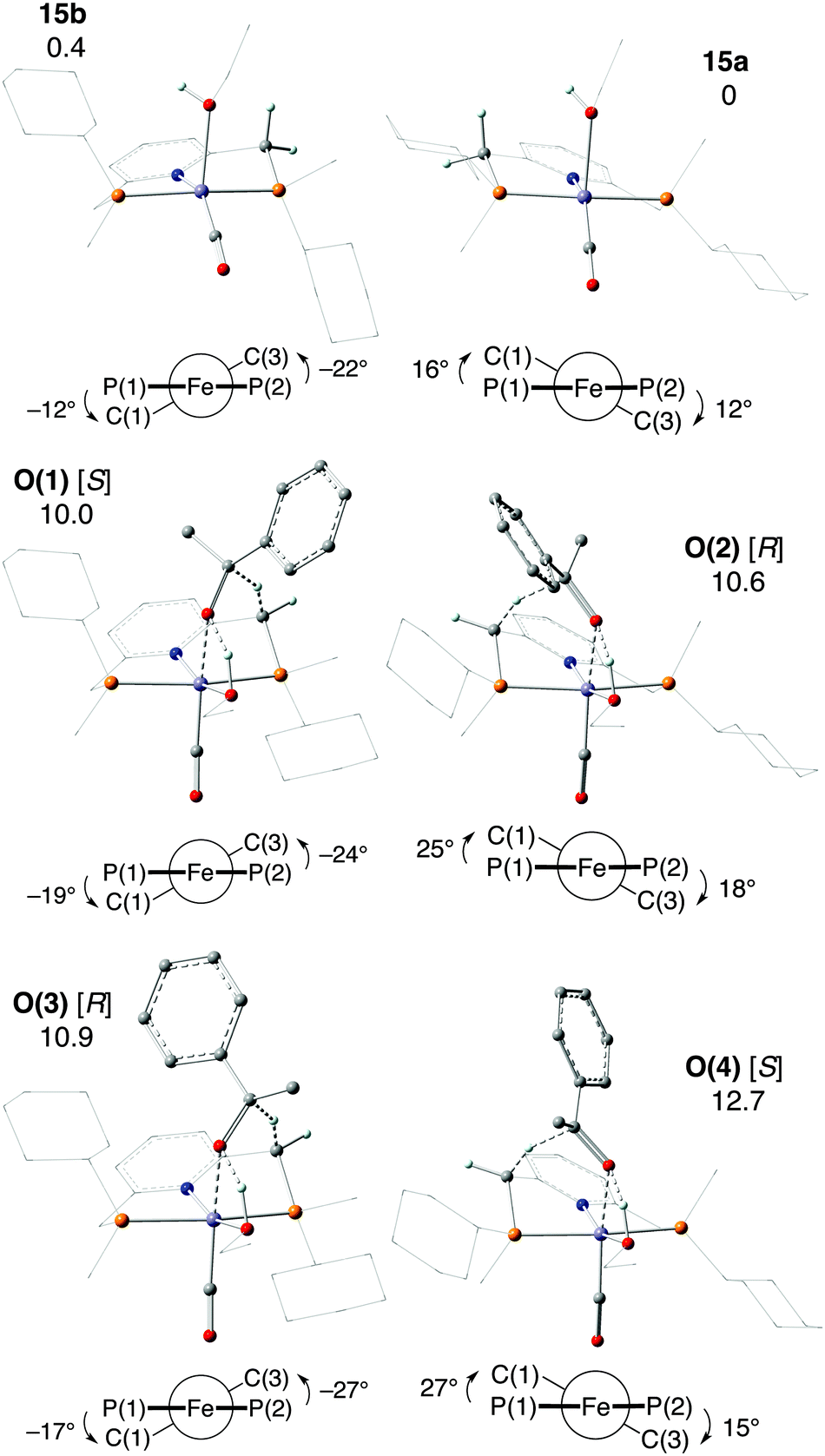 | ||
| Fig. 3 Iron(0) complexes 15a and 15b and TSs O(1)–O(4) for hydride transfer in mechanism O (product stereochemistry in brackets). Newman projections give θ1 (left) and θ2 (right). Relative Gibbs free energies are in kcal mol−1 (reprinted with permission from ref. 88. Copyright 2018 American Chemical Society). | ||
The intrinsic reaction coordinate (IRC) analysis shows that O(2) and O(4) connect to 15a, which bears equatorial cyclohexyl groups, whereas O(1) and O(3) are linked to 15b, which bears axial cyclohexyl groups. Also, conformer 15a, which bears equatorial cyclohexyl substituents, converts to 15b, which is less stable by 0.4 kcal mol−1.
Conformers 15a and 15b interconvert rapidly and react under Curtin–Hammett conditions to give the 1-phenylethoxide complexes 16a/16a′ and 16b/16b′ (Fig. 4). This implies that the ratio of the products depends on the energy difference between the transition states.98
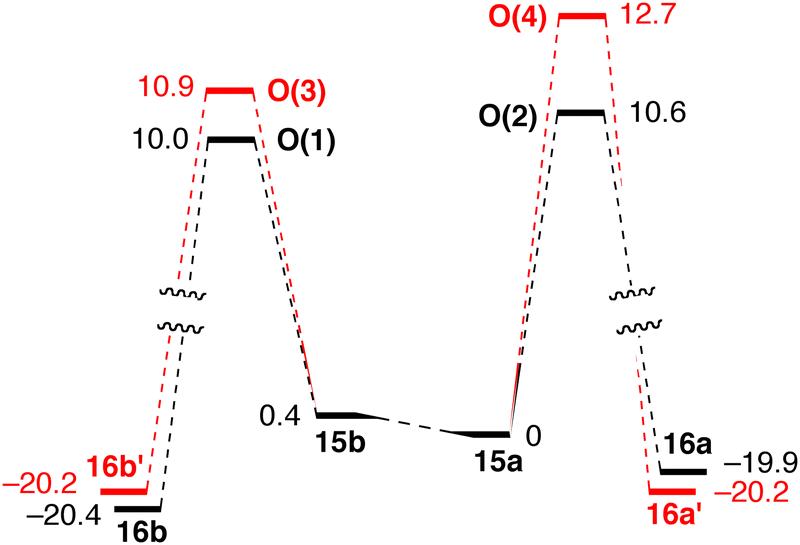 | ||
| Fig. 4 Reaction pathway for H− transfer in mechanism O. Relative Gibbs free energies are in kcal mol−1 (reprinted with permission from ref. 88. Copyright 2018 American Chemical Society). | ||
The small changes in |θ| (Fig. 3) indicate minor structural reorganization between the Fe(0) species 15a/15b and O(1)–O(4), which further supports the retention of conformation along the reaction coordinate. Conformer 15b (with axial cyclohexyl groups) is just 0.4 kcal mol−1 less stable than 15a, which bears equatorial cyclohexyls. The major contribution to the total energy of the O(1)–O(4) transition states is the distortion of the catalyst, i.e. the conformational change of the chelate ring involved in hydride transfer. However, the energetic preference for conformations in which the large P-substituents occupy equatorial positions is small, and can be reversed as the substrate approaches. Even weak intermolecular interactions, such as C–H⋯O or C–H⋯π, swap the relative stability of the conformations of different intermediates and transition states. Therefore, it is reasonable to assume that the conformational flexibility observed for 1a might be a more general problem with P-stereogenic pincer ligands with a pyridine backbone (see below).
Comparison with other P-stereogenic PNP/PCP ligands
PNP and PCP ligands similar to ligand 1a have been studied for their application in hydrogenation (Scheme 31)84,85c,86c and hydrophosphination/hydroamination (Scheme 32).85b,d,e,87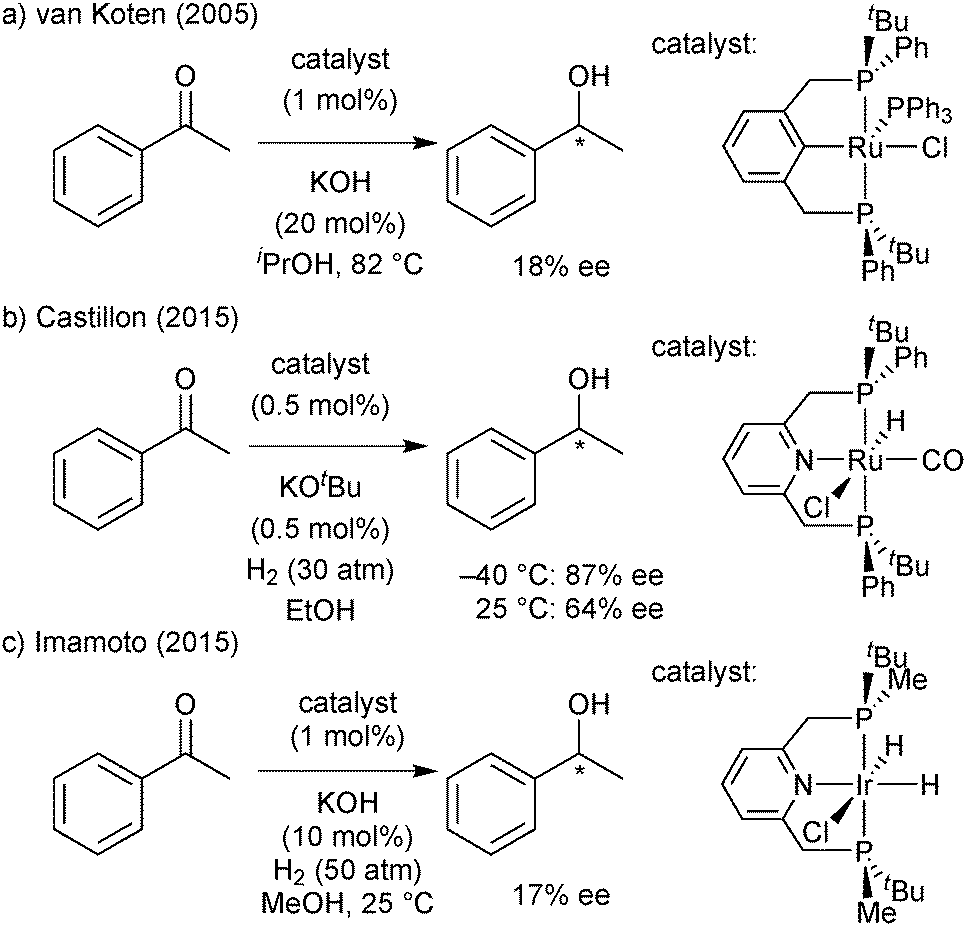 | ||
| Scheme 31 PNP/PCP ligands in hydrogenation reactions. | ||
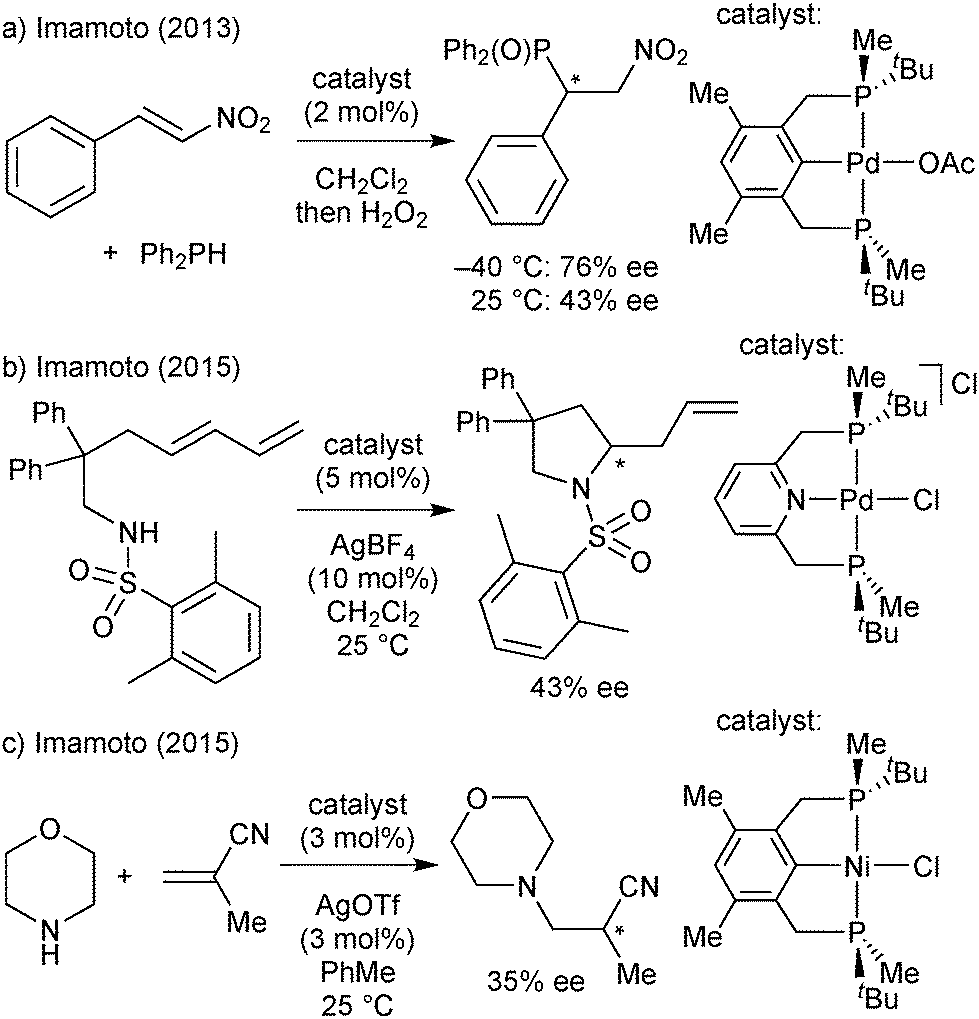 | ||
| Scheme 32 PNP/PCP ligands in hydrophosphination and hydroamination reactions. | ||
Overall, the enantioselectivity was modest and/or strongly temperature dependent. In the ruthenium-catalysed ketone hydrogenation of ketones, the reduction of acetophenone was much more enantioselective at −40 than at 25 °C (87 and 64% ee, respectively, Scheme 31b).84 Palladium-catalysed hydrophosphinations show similar effects of the temperature (Scheme 32a, ref. 85b gives a further example).85b,87
Conformational equilibria of the 2,6-dimethylenepyridine (PNP) or 1,3-xylyl (PCP) backbones analogous to those described above for 11a may account for these temperature effects, but a conformational analysis would be needed to verify this hypothesis. Still, our observations with the PNP pincer ligand 1a prompted us to move to PN(H)P ligands with N,N-diethyleneamine as a backbone, in which the sp3 nitrogen atom should act as a conformational lock upon coordination to a metal centre.
PN(H)P pincer complexes
Synthesis of PN(H)P pincers
As for their PNP analogues, we prepared the borane-protected PN(H)P pincer ligands 17a–17c by deprotonation of 3a and 3b and subsequent alkylation with bis(2-chloroethyl)-N-trimethylsilylamine (Scheme 33).90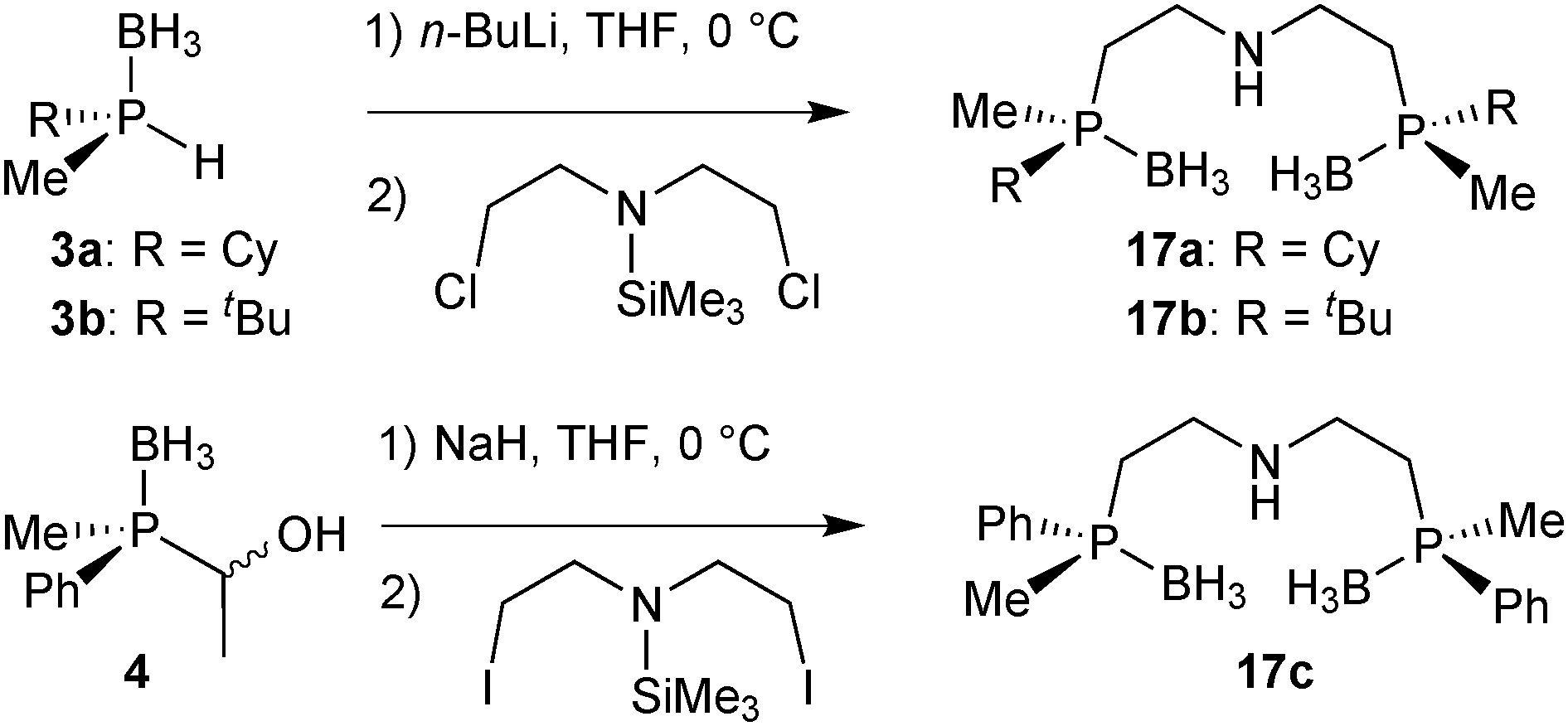 | ||
| Scheme 33 Alkylation of secondary phosphine boranes to give the protected PN(H)P pincers. | ||
The phenylmethylphosphine derived pincer 17c was prepared from the masked secondary phosphine borane 4 and bis(2-iodoethyl)-N-trimethylsilylamine (analogously to 9c). The borane-protected pincer ligands 17a–17c are indefinitely stable upon storage in air at room temperature.
[FeBr2(CO)(PN(H)P)]
After deprotection of 17a–17c with HBF4·OEt2 in dichloromethane,72 pincers 2a–2c form the dibromocarbonyl complexes [FeBr2(CO)(2a–2c)] (18a–18c) by reaction with [FeBr2(PPh3)2] under a CO atmosphere (Scheme 34). Complexes 18a and 18c are blue with a meridional pincer ligand and mutually trans bromides, whereas the tert-butylmethylphosphine-derivative 5b is red with facial pincer and cis bromides.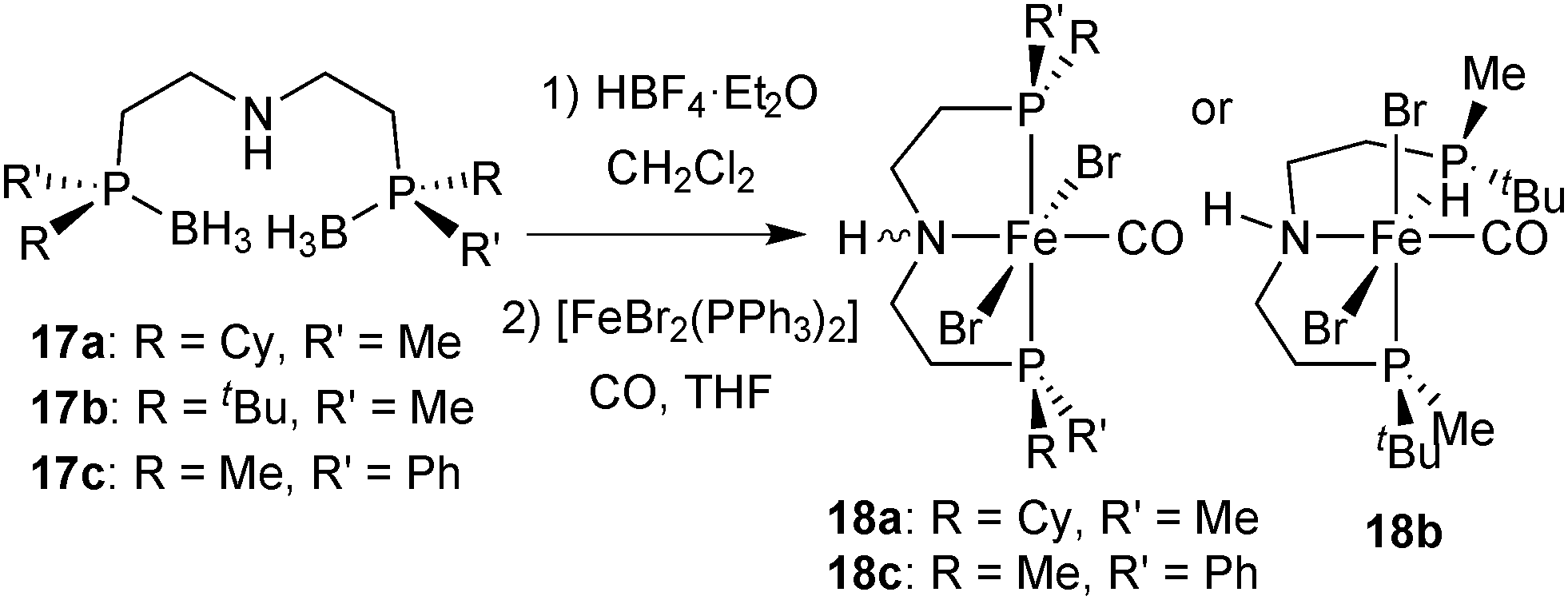 | ||
| Scheme 34 Synthesis of dibromocarbonyl complexes 5a–5c. | ||
Overall, the PN(H)P pincer ligands 2a–2c form iron complexes that are less stable than those with the pyridine backbone (1a–1c). Hence, complexes 18a–18c are best handled under an inert atmosphere, although brief exposure to ambient conditions is tolerated. In particular, the tert-butylmethylphosphine derivative 18b is unstable and slowly decomposes both in solution and in the solid state.
The conformation of the chelate rings with the N,N-diethylamine backbone is rigidified by the sp3 nitrogen (Fig. 5). The metallacycles adopt an envelope conformation with C(2) and C(3) in the endo position, and the elongated ellipsoids of the backbone indicate the presence of an isomeric form where the N–H proton points downwards, leading to a ring flip.
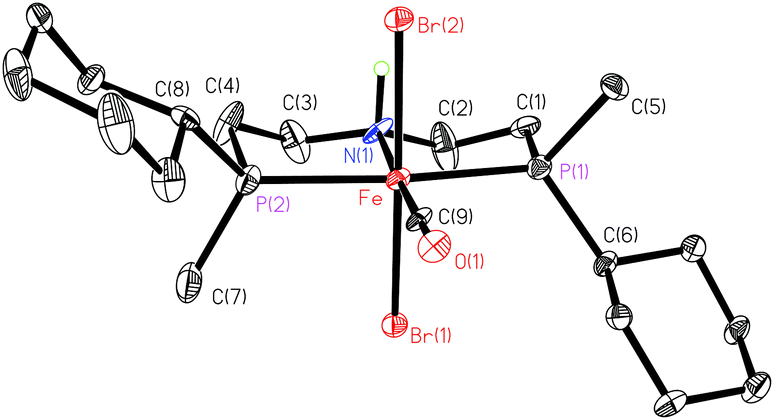 | ||
| Fig. 5 ORTEP drawing of 18a (reprinted with permission from ref. 90). | ||
Hydride complexes
Treatment of the dibromocarbonyl complexes 18a–18c with NaBHEt3 (1 equiv.) in THF or CH2Cl2 gave the bromohydridocarbonyl derivatives 19a–19c (Scheme 35).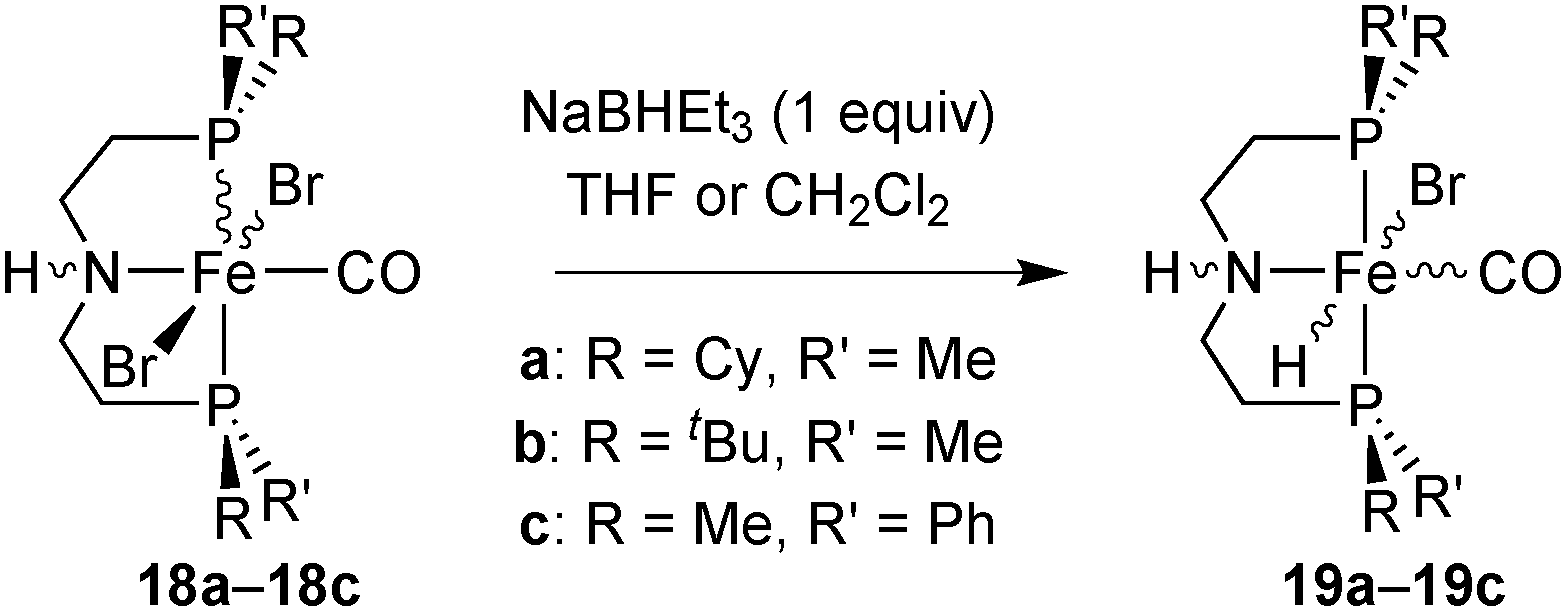 | ||
| Scheme 35 Synthesis of PN(H)P bromohydridocarbonyl complexes. | ||
The hydride ligand is located trans to bromide in the major isomer of hydride 19a and trans to CO in 19b and 19c. Hydrides 19a–19c are extremely sensitive towards air and moisture. As they decompose in solution, they were freshly prepared for each catalytic run.
Hydrides 19a–19c as catalysts
In a preliminary screening, we tested hydrides 19a and 19b in the hydrogenation of acetophenone (13) after dehydrobromination.90 The P(Cy)Me analogue 19a gave the best activity in toluene (quantitative conversion after 16 h at 5.5 bar H2), but with low enantioselectivity (19% ee), and the P(tBu)Me derivative 19b was not active (Scheme 36).90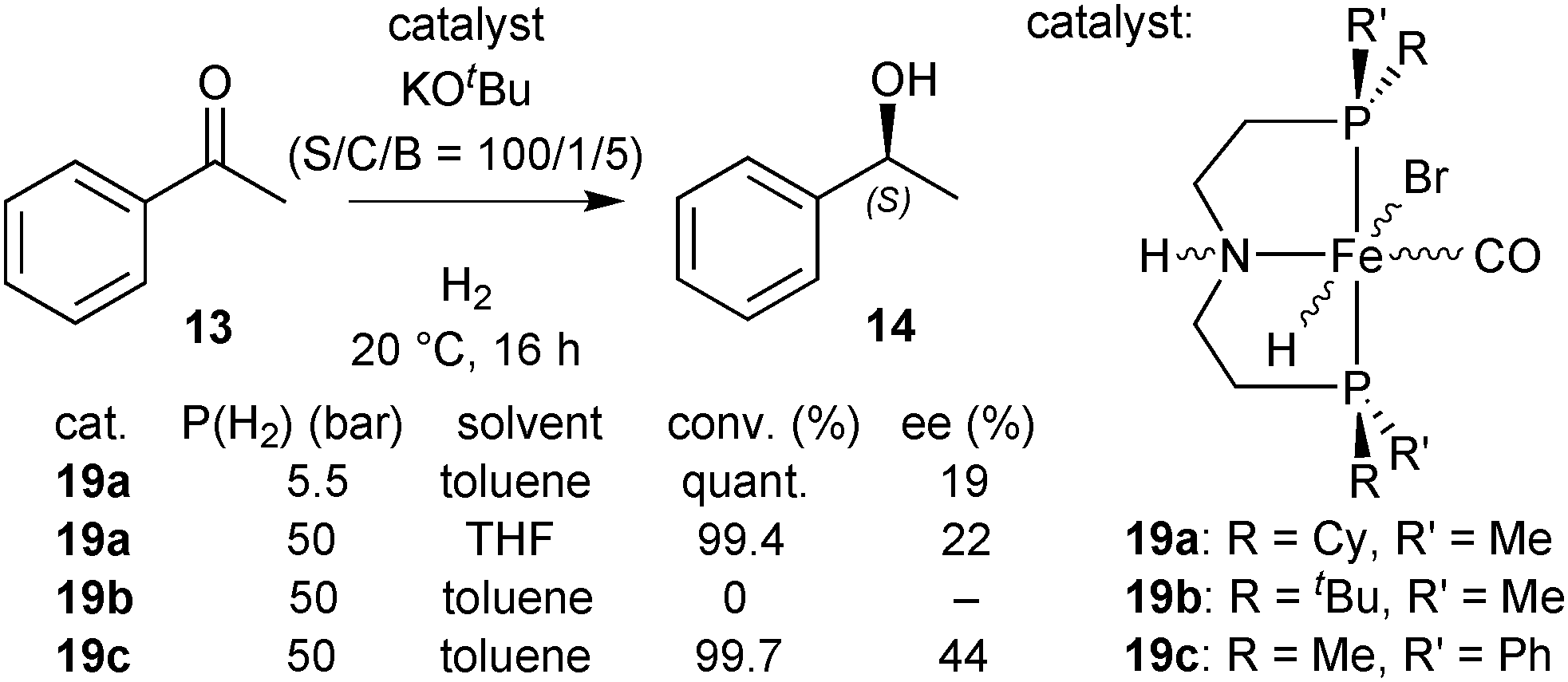 | ||
| Scheme 36 Asymmetric hydrogenation of acetophenone with 19a–19c. | ||
Origin of enantioselectivity with Fe/PN(H)P catalysts
To explain the low enantioselectivity of 19a and improve the ligand design, we used DFT calculations to model the enantiodetermining hydride transfer step TS20a, assuming the same mechanism as the achiral [FeBrH(CO)(iPr2P(CH2)2)N(H)(CH2)2PiPr2]21b (Scheme 37).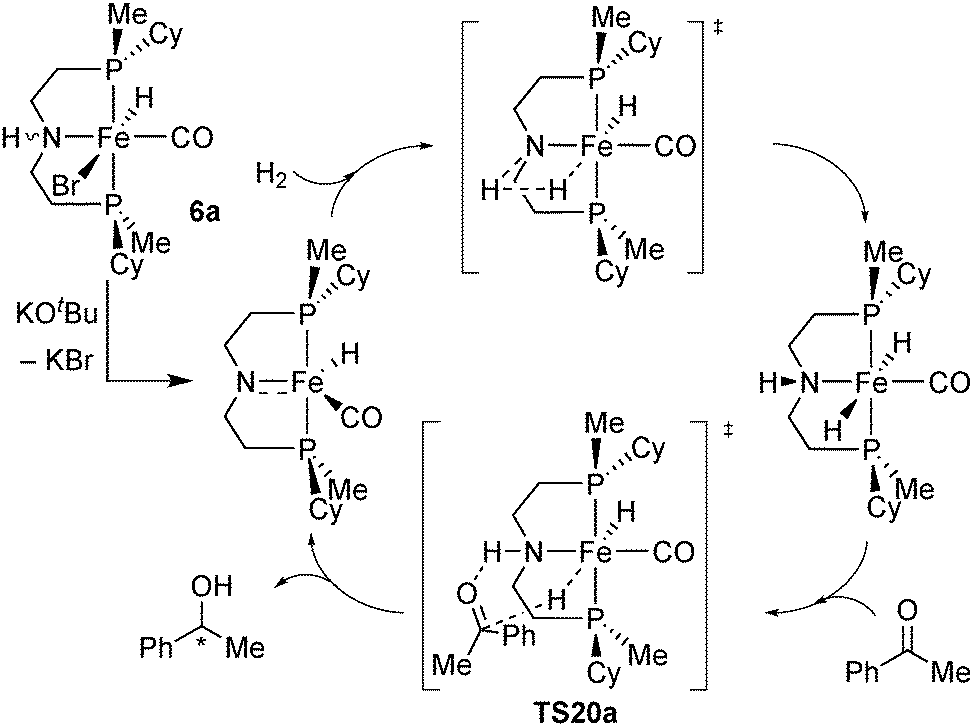 | ||
| Scheme 37 Proposed mechanism for the hydrogenation of ketones with Fe/PN(H)P catalyst 31a. | ||
The diastereoisomeric transition states TS20aS and TS20aR lead to (S)- and (R)-1-phenylethanol, respectively (Fig. 6 top). The calculated energy difference (0.5 kcal mol−1, hybrid DFT model at the B3LYP level of theory) is in fair agreement with the experimental one (ca. 0.2 kcal mol−1).
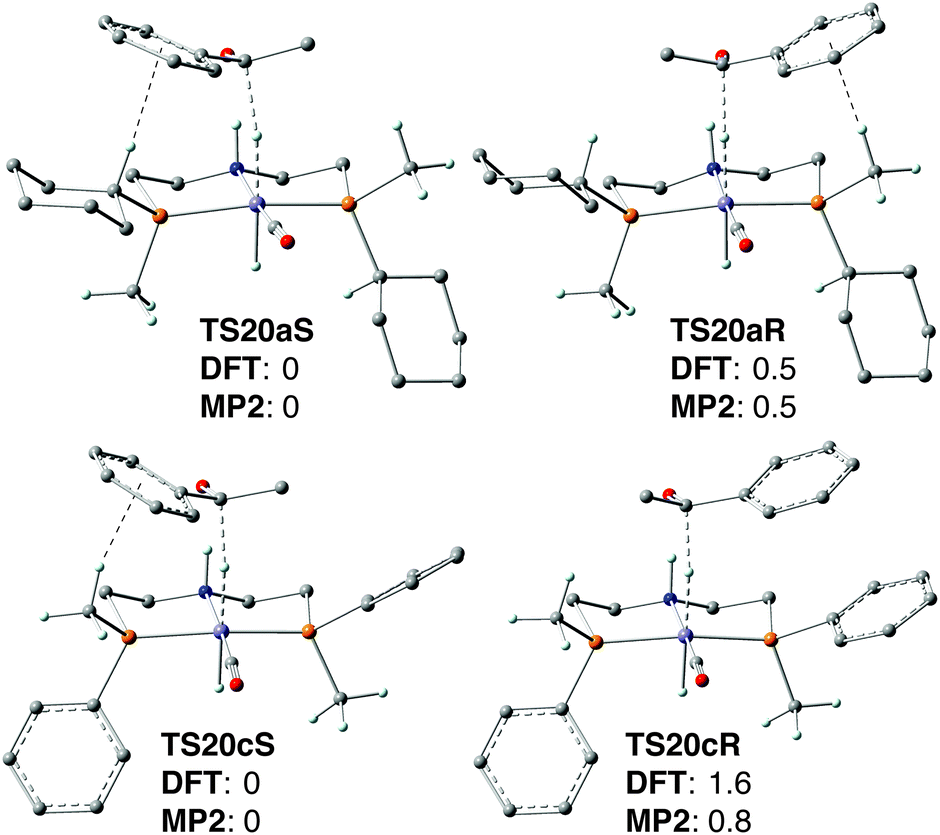 | ||
| Fig. 6 Transition states TS20a/cS and TS20a/cR for the hydride transfer. Relative Gibbs free energies are in kcal mol−1. Black dashed lines depict the hydride transfer and the C–H⋯π interaction. | ||
We speculated that the low energy difference between TS20aS and TS20aR may result from the occurrence of a similar CH/π interaction with the phenyl group of acetophenone in both transition states which involves the Cipso–H of the cyclohexyl in TS20aS and the methyl on phosphorus in TS20aR (Fig. 6 top) (in addition to a strong N–H⋯O hydrogen bond that directs and activates acetophenone). Therefore, we tried to increase the enantioselectivity by eliminating selectively one of CH/π interactions.
To this goal, the cyclohexyl group was replaced with a phenyl ring in silico, which eliminated the C–H⋯π interaction in transition state TS20cR (Fig. 6 bottom). This gave an increased energy difference of 1.6 kcal mol−1 between the diastereoisomeric TSs TS20cS and TS20cR, which corresponds to 88% ee. These computational results motivated us to tackle the synthetic challenge of introducing an enantiomerically pure phenylmethylphosphine unit onto the backbone (a primary alkyl halide) and prepare the new pincer ligand 2c as described above.
Disappointingly, however, the phenylmethylphosphine-derived bromocarbonylhydride complex [FeBrH(CO)(2c)] (19c) gave only 44% ee (Scheme 36), which implies an energy difference for the enantiodetermining transition states of 0.6 kcal mol−1. Surprised at first, we tested other DFT functionals and basis sets, but all gave energy values within ±0.4 kcal mol−1 of 1.6 kcal mol−1. Eventually, single-point energy calculations with MP2 gave an energy difference of 0.6 kcal mol−1 between TS20cS and TS20cR (Fig. 6 bottom), which perfectly corresponds to the experimental enantioselectivity of 44% ee. Most probably, DFT fails to predict the enantioselectivity accurately with the P(Me)Ph-based pincer 2c because it overestimates the energy of the C–H⋯π interactions, which is a known effect.90 When the diastereoisomeric TSs contain the same number of CH/π interactions (such as TS20aS and TS20aR, which have one each, Fig. 6), the errors cancel out, and DFT and MP2 give the same result. Instead, for 2c, only the less energetic TS20cS features a CH/π interaction whose energy is most probably overestimated by DFT, leading to a spuriously high energy difference with TS20cR (1.6 kcal mol−1, vs. 0.8 kcal mol−1 by MP2).
A further intriguing feature is that, although catalysts 19a and 19c contain pincer ligands with opposite P-stereochemistry (S vs. R, respectively) they hydrogenate acetophenone with the same sense of induction (S). Therefore, the methyl group acts as the “small” substituent with PMe(Ph) (TS20c), but as the “large” one with P(Cy)Me (TS20a) (Fig. 6), which suggests that attractive non-covalent interactions (NCI, e.g. CH/π, π/π stacking), rather than repulsive steric hindrance, dominate (or at least strongly influence) enantioselection with these ligands, which are not exceedingly bulky. The NCI analysis supports this view.90 Interestingly, similar observations have been made with P-stereogenic diphosphines in the context of the quadrant rule. Gridnev has identified attractive interactions (C–H/π or C–H/H–C) between the coordinated substrate and the ligand even in the crowded quadrants. Increasing the overall steric bulk tightens the contacts and turns attraction to repulsion.35b
The importance of non-covalent interactions poses a further challenge to the rational ligand design by calculation. In particular, hybrid DFT methods, although a reliable tool to study the geometry of transition states, do not deliver energy values that are accurate enough to predict the enantioselectivity when weak interactions (such as C–H⋯π) are involved. Post Hartree–Fock methods such as MP2 are more accurate, but much more demanding computationally than hybrid DFT methods.
Conclusions and outlook
Differences between dialkyl- and diarylphosphine donors
When we started our study of P-stereogenic pincer ligands some years back, we chose dialkylphosphine donors by analogy with the existing achiral PNP pincer complexes of iron(II). In particular, the combination of methyl with a bulky group such as tert-butyl or cyclohexyl, as in Imamoto's approach, seemed promising. In retrospect, we recognise now that PR2 based ligands are more difficult to design than those based on PAr2 donors (R = alkyl, Ar = aryl). The above sections have shown that conformational issues are (literally) pivotal in P-stereogenic pincer ligands as well as in bidentate ones.Thus, in P-stereogenic diphosphines, the substituents of phosphorus must differ largely in size to lock the chelate in the conformation in which the large substituent occupies the equatorial position. As aryl substituents on phosphorus are flat, their apparent size depends on their conformation. Hence, in DiPAMP, the axial phenyl group acts as the small substituent with respect to the chelate, but as the large substituent toward the substrate (Scheme 6). With P(tBu)Me (or P(Cy)Me) as donors, instead, the large substituent occupies the equatorial position, leaving the small methyl group axial. In this conformation, both substituents act as small, notwithstanding from which direction the substrate approaches. This is illustrated by the crystal structure of 18a (Fig. 5) and in the transition state TS20aS (Fig. 6). Therefore, one might speculate that methyl as the axial group is too small to enforce strong enantiodiscrimination. In fact, the situation is more complex, as our studies with the PNP and PN(H) pincers show.
Overall steric bulk and attractive interactions
We have found that, in the enantiodetermining transition states, the PNP pincer ligand with P(Cy)Me donors (1a) can favour the conformation that features axial cyclohexyl groups (Fig. 3). This obviously results from the flexibility of the backbone that allows for the axial/equatorial switch, but also suggests that the overall steric bulk on the pincer is not very large (otherwise the axial cyclohexyl would cost too much energy), at least in outer sphere reactions as those studied here. The modest enantioselectivity achieved with the conformationally rigid PN(H)P pincer with P(Ph)Me donors (2c, Scheme 36) supports the view that methyl-containing P-stereogenic units do not offer adequate steric bulk. In this context, the DFT study with the PN(H)P pincer ligand 2c indicates that attractive interactions play a major role in enantiodiscrimination. This may be seen as an indication that, when steric repulsions are moderate, attractive non-covalent interactions also play a significant role in enantiodiscrimination as already discussed for P-stereogenic diphosphines.35b We recognise that our understanding of the mechanisms of chirality transfer is, even after so many decades, still fragmentary and incomplete. DFT studies are increasingly important in this respect, but their accuracy in the descriptions of weak interactions needs improvement before rational ligand design by calculation becomes reality in asymmetric catalysis.Outlook
Overall, the topic of this article is the use of stereogenic phosphorus to control the conformation of a chelate, be it in a bidentate or in a tridentate ligand. We have discussed the pitfalls of this approach at length, and concluded that a rigid backbone should be used in such ligands. Obviously, the straightforward method to solve it is to incorporate a stereocentre in the backbone. This does not necessarily mean that purely P-stereogenic pincer ligands have little chance of success, though, and does not relegate stereogenic phosphorus to a mere supporting role. In particular, the combination of P-stereogenic diarylphosphine units (PArAr′) with dialkyl ones (even achiral such as PR2), which is unexplored yet, might increase the overall steric bulk and hence enantioface discrimination without decreasing significantly the stability of the catalyst. In fact, iron(II) complexes with mixed PiPr/PPh2 donors have been prepared and used in asymmetric hydrogenation (Scheme 2).26–28Alternatively stereogenic phosphorus can be exploited to direct the configuration of multiple chelate rings of a polydentate ligand. In this case, the multichelate structure is the element of rigidity whose necessity has been recognised in the above discussion. With our N2P2 macrocyclic ligands, we have shown that this approach generates extremely rigid structures with predictable structure and absolute configuration that deliver high enantioselectivity in the transfer hydrogenation of ketones for a broad substrate scope.10 Therefore, we believe that stereogenic phosphorus will continue to play an important role in the synthesis and stereochemical understanding of asymmetric catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to ETH Zürich (grant ETH-36 17-1) and to the Swiss National Science Foundation (grant 200020-146881) for financial support.Notes and references
- (a) A. Grabulosa, P-Stereogenic Ligands in Enantioselective Catalysis, RSC Publishing, Cambridge, 2011 Search PubMed; (b) M. Dutartre, J. Bayardon and S. Jugé, Chem. Soc. Rev., 2016, 45, 5771 RSC.
- Non-Noble Metal Catalysis: Molecular Approaches and Reactions, ed. R. J. Klein Gebbink and M. C. Moret, Wiley-VCH, Weinheim, 2019 Search PubMed.
- I. Bauer and H. J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed.
- A. Fürstner, Angew. Chem., Int. Ed., 2009, 48, 1364 CrossRef.
- The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007 Search PubMed.
- H. U. Blaser, B. Pugin and F. Spindler, Top. Organomet. Chem., 2012, 42, 65 CrossRef CAS.
- Seminal papers: (a) T. Ohkuma, H. Ooka, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 2675 CrossRef CAS; (b) J. X. Gao, T. Ikariya and R. Noyori, Organometallics, 1996, 15, 1087 CrossRef CAS . For accounts of the evolution from ruthenium to iron, see: ; (c) R. H. Morris, Chem. Rec., 2016, 16, 2644 CrossRef; (d) R. H. Morris, Dalton Trans., 2018, 47, 10809 RSC.
- C. Sui-Seng, F. Freutel, A. J. Lough and R. H. Morris, Angew. Chem., Int. Ed., 2008, 47, 940 CrossRef CAS.
- W. Zuo, A. J. Lough, Y. F. Liand and R. H. Morris, Science, 2013, 342, 1080 CrossRef CAS.
- Selected papers: (a) R. Bigler, R. Huber and A. Mezzetti, Angew. Chem., Int. Ed., 2015, 54, 5171 CrossRef CAS; (b) L. De Luca and A. Mezzetti, Angew. Chem., Int. Ed., 2017, 56, 11949 CrossRef CAS; (c) L. De Luca, A. Passera and A. Mezzetti, J. Am. Chem. Soc., 2019, 141, 2545 CrossRef.
- V. Rautenstrauch, X. Hoang-Cong, R. Churlaud, K. Abdur-Rashid and R. H. Morris, Chem. – Eur. J., 2003, 9, 4954 CrossRef CAS.
- W. W. Zuo, S. Tauer, D. E. Prokopchuk and R. H. Morris, Organometallics, 2014, 33, 5791 CrossRef CAS.
- S. Werkmeister, J. Neumann, K. Junge and M. Beller, Chem. – Eur. J., 2015, 21, 12226 CrossRef CAS.
- (a) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979 CrossRef CAS; (b) K. L. Szabo and O. F. Wendt, Pincer and Pincer-Type Complexes: Applications in Organic Synthesis and Catalysis, Wiley-VCH, Weinheim, 2014 CrossRef.
- J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832 CrossRef CAS.
- R. Langer, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 2120 CrossRef CAS.
- T. Zell, Y. Ben-David and D. Milstein, Catal. Sci. Technol., 2015, 5, 822 RSC.
- J. A. Garg, S. Chakraborty, Y. Ben-David and D. Milstein, Chem. Commun., 2016, 52, 5285 RSC.
- T. Zell, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2014, 53, 4685 CrossRef CAS.
- N. Gorgas, B. Stöger, L. F. Veiros, E. Pittenauer, G. Allmaier and K. Kirchner, Organometallics, 2014, 33, 6905 CrossRef CAS.
- (a) S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. J. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722 CrossRef CAS . See also: ; (b) H. Jiao, K. Junge, E. Alberico and M. Beller, J. Comput. Chem., 2016, 37, 168 CrossRef CAS.
- S. Chakraborty, H. G. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and H. R. Guan, J. Am. Chem. Soc., 2014, 136, 7869 CrossRef CAS.
- (a) K. J. Haack, S. Hashiguchi, A. Fujii, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 285 CrossRef CAS; (b) R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931 CrossRef CAS; (c) T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300 CrossRef CAS; (d) T. Ikariya, Bull. Chem. Soc. Jpn., 2011, 84, 1 CrossRef CAS; (e) P. A. Dub and J. C. Gordon, Dalton Trans., 2016, 45, 6756 RSC.
- S. Chakraborty, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2014, 136, 8564 CrossRef CAS PubMed.
- S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen, W. D. Jones and S. Schneider, ACS Catal., 2014, 4, 3994 CrossRef CAS.
- P. O. Lagaditis, P. E. Sues, J. F. Sonnenberg, K. Y. Wan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2014, 136, 1367 CrossRef CAS.
- S. A. M. Smith, P. O. Lagaditis, A. Lüpke, A. J. Lough and R. H. Morris, Chem. – Eur. J., 2017, 23, 7212 CrossRef CAS.
- A. Zirakzadeh, K. Kirchner, A. Roller, B. Stöger, M. Widhalm and R. H. Morris, Organometallics, 2016, 35, 3781 CrossRef CAS.
- H. U. Blaser, Chem. Rev., 1992, 92, 935 CrossRef CAS.
- M. D. Fryzuk and B. Bosnich, J. Am. Chem. Soc., 1977, 99, 6262 CrossRef CAS.
- R. S. Cahn, C. Ingold and V. Prelog, Angew. Chem., 1966, 78, 413 CrossRef.
- (a) H. Brunner, A. Winter and J. Breu, J. Organomet. Chem., 1998, 553, 285 CrossRef CAS . For definitions, see: ; (b) D. Cremer, Isr. J. Chem., 1980, 20, 12 CrossRef CAS.
- (a) W. S. Knowles, M. J. Sabacky, B. D. Vineyard and D. J. Weinkauff, J. Am. Chem. Soc., 1975, 97, 2567 CrossRef CAS; (b) B. D. Vineyard, W. S. Knowles, M. J. Sabacky, G. L. Bachman and D. J. Weinkauff, J. Am. Chem. Soc., 1977, 99, 5946 CrossRef CAS.
- For early studies, see: C. L. Landis and J. Halpern, J. Am. Chem. Soc., 1987, 109, 1746 CrossRef CAS , and references therein.
- (a) I. D. Gridnev and T. Imamoto, Acc. Chem. Res., 2004, 37, 633 CrossRef CAS . For a discussion of attraction vs. repulsion, see: ; (b) I. D. Gridnev, ChemCatChem, 2016, 8, 3463 CrossRef CAS.
- J. S. Giovannetti, C. M. Kelly and C. R. Landis, J. Am. Chem. Soc., 1993, 115, 4040 CrossRef CAS.
- W. S. Knowles, Acc. Chem. Res., 1983, 16, 106 CrossRef CAS.
- H. Tsuruta, T. Imamoto, K. Yamaguchi and I. D. Gridnev, Tetrahedron Lett., 2005, 46, 2879 CrossRef CAS.
- (a) B. Zupancic, B. Mohar and M. Stephan, Adv. Synth. Catal., 2008, 350, 2024 CrossRef CAS; (b) M. Stephan, D. Šterk and B. Mohar, Adv. Synth. Catal., 2009, 351, 2779 CrossRef CAS; (c) B. Zupančič, B. Mohar and M. Stephan, Org. Lett., 2010, 12, 1296 CrossRef.
- F. Maienza, F. Spindler, M. Thommen, B. Pugin, C. Malan and A. Mezzetti, J. Org. Chem., 2002, 67, 5239 CrossRef CAS.
- Asymmetric hydrogenation of functionalised olefins: (a) T. Imamoto, J. Watanabe, Y. Wada, H. Masuda, H. Yamada, H. Tsuruta, S. Matsukawa and K. Yamaguchi, J. Am. Chem. Soc., 1998, 120, 1635 CrossRef CAS; (b) A. Ohashi and T. Imamoto, Org. Lett., 2001, 3, 373 CrossRef CAS; (c) A. Ohashi, S. I. Kikuchi, M. Yasutake and T. Imamoto, Eur. J. Org. Chem., 2002, 2535 CrossRef CAS . Arylation of enones: ; (d) T. Imamoto, Y. Saitoh, A. Koide, T. Ogura and K. Yoshida, Angew. Chem., Int. Ed., 2007, 46, 8636 CrossRef CAS . Iridium-catalysed imine hydrogenation: ; (e) T. Imamoto, N. Iwadate and K. Yoshida, Org. Lett., 2006, 8, 2289 CrossRef CAS . Ruthenium-catalysed asymmetric hydrogenation of β-ketoesters: ; (f) T. Yamano, N. Taya, M. Kawada, T. Huang and T. Imamoto, Tetrahedron Lett., 1999, 40, 2577 CrossRef CAS . Pd-catalysed allylic alkylation: ; (g) N. Oohara, K. Katagiri and T. Imamoto, Tetrahedron: Asymmetry, 2003, 14, 2171 CrossRef CAS.
- I. D. Gridnev, Y. Yamanoi, N. Higashi, H. Tsuruta, M. Yasutake and T. Imamoto, Adv. Synth. Catal., 2001, 343, 118 CrossRef CAS.
- Y. Yamanoi and T. Imamoto, J. Org. Chem., 1999, 64, 2988 CrossRef CAS.
- I. D. Gridnev, M. Yasutake, N. Higashi and T. Imamoto, J. Am. Chem. Soc., 2001, 123, 5268 CrossRef CAS.
- (a) G. Hoge, H. P. Wu, W. S. Kissel, D. A. Pflum, D. J. Greene and J. Bao, J. Am. Chem. Soc., 2004, 126, 5966 CrossRef CAS; (b) H. P. Wu and G. Hoge, Org. Lett., 2004, 6, 3645 CrossRef CAS PubMed; (c) I. D. Gridnev, T. Imamoto, G. Hoge, M. Kouchi and H. Takahashi, J. Am. Chem. Soc., 2008, 130, 2560 CrossRef CAS PubMed.
- (a) E. Cristobal-Lecina, P. Etayo, S. Doran, M. Revés, P. Martin-Gago, A. Grabulosa, A. R. Costantino, A. Vidal-Ferran, A. Riera and X. Verdaguer, Adv. Synth. Catal., 2014, 356, 795 CrossRef CAS; (b) E. Cristobal-Lecina, A. R. Costantino, A. Grabulosa, A. Riera and X. Verdaguer, Organometallics, 2015, 34, 4989 CrossRef CAS.
- K. Mislow and J. Siegel, J. Am. Chem. Soc., 1984, 106, 3319 CrossRef CAS.
- (a) M. J. Burk, J. E. Feaster, W. A. Nugent and R. L. Harlow, J. Am. Chem. Soc., 1993, 115, 10125 CrossRef CAS . See also: ; (b) S. Rast, M. Stephan and B. Mohar, Eur. J. Org. Chem., 2015, 2214 CrossRef CAS.
- (a) T. Miura and T. Imamoto, Tetrahedron Lett., 1999, 40, 4833 CrossRef CAS; (b) K. Tamura, M. Sugiya, K. Yoshida, A. Yanagisawa and T. Imamoto, Org. Lett., 2010, 12, 4400 CrossRef CAS.
- T. Imamoto, K. Sugita and K. Yoshida, J. Am. Chem. Soc., 2005, 127, 11934 CrossRef CAS.
- Functionalised alkenes: (a) T. Imamoto, K. Tamura, Z. Zhang, Y. Horiuchi, M. Sugiya, K. Yoshida, A. Yanagisawa and I. D. Gridnev, J. Am. Chem. Soc., 2012, 134, 1754 CrossRef CAS . Functionalised α-dehydroamino ketones: ; (b) Q. Hu, J. Chen, Z. Zhang, Y. Liu and W. Zhang, Org. Lett., 2016, 18, 1290 CrossRef CAS PubMed . Allylic hydrazones: ; (c) Q. P. Hu, Y. H. Hu, Y. Liu, Z. F. Zhang, Y. G. Liu and W. B. Zhang, Chem. – Eur. J., 2017, 23, 1040 CrossRef CAS.
- Y. Shibata and K. Tanaka, J. Am. Chem. Soc., 2009, 131, 12552 CrossRef CAS PubMed.
- I. H. Chen, L. Yin, W. Itano, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 11664 CrossRef CAS.
- (a) K. Kubota, E. Yamamoto and H. Ito, Adv. Synth. Catal., 2013, 355, 3527 CrossRef CAS; (b) E. Yamamoto, Y. Takenouchi, T. Ozaki, T. Miya and H. Ito, J. Am. Chem. Soc., 2014, 136, 16515 CrossRef CAS; (c) K. Kubota, Y. Watanabe and H. Ito, Adv. Synth. Catal., 2016, 358, 2379 CrossRef CAS.
- (a) K. Yao, D. Liu, Q. Yuan, T. Imamoto, Y. Liu and W. Zhang, Org. Lett., 2016, 18, 6296 CrossRef CAS; (b) T. Imamoto, M. Nishimura, A. Koide and K. Yoshida, J. Org. Chem., 2007, 72, 7413 CrossRef CAS.
- M. Jin, L. Adak and M. Nakamura, J. Am. Chem. Soc., 2015, 137, 7128 CrossRef CAS.
- S. I. Featherman and L. D. Quin, J. Am. Chem. Soc., 1975, 97, 4349 CrossRef CAS.
- C. R. Johnson and T. Imamoto, J. Org. Chem., 1987, 52, 2170 CrossRef CAS.
- T. Imamoto, Y. Horiuchi, E. Hamanishi, S. Takeshita, K. Tamura, M. Sugiya and K. Yoshida, Tetrahedron, 2015, 71, 6471 CrossRef CAS.
- J. Bakos, I. Toth, B. Heil, G. Szalontai, L. Parkanyi and V. Fülöp, J. Organomet. Chem., 1989, 370, 263 CrossRef CAS.
- R. M. Stoop, F. Spindler and A. Mezzetti, Organometallics, 1998, 17, 668 CrossRef CAS.
- J. Holz, K. Rumpel, A. Spannenberg, R. Paciello, H. J. Jiao and A. Börner, ACS Catal., 2017, 7, 6162 CrossRef CAS.
- (a) U. Nettekoven, P. C. J. Kamer, P. W. N. M. van Leeuwen, M. Widhalm, A. L. Spek and M. Lutz, J. Org. Chem., 1999, 64, 3996 CrossRef CAS; (b) F. Maienza, M. Wörle, P. Steffanut, A. Mezzetti and F. Spindler, Organometallics, 1999, 18, 1041 CrossRef CAS.
- E. Salomó, A. Gallen, G. Sciortino, G. Ujaque, A. Grabulosa, A. Lledós, A. Riera and X. Verdaguer, J. Am. Chem. Soc., 2018, 140, 16967 CrossRef.
- (a) S. Jugé, M. Stephan, J. A. Laffitte and J. P. Genêt, Tetrahedron Lett., 1990, 31, 6357 CrossRef; (b) S. Jugé, M. Stephan, R. Merdès, J. P. Genêt and S. Hault-Desportes, J. Chem. Soc., Chem. Commun., 1993, 531 RSC; (c) S. Jugé, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 233 CrossRef.
- A. R. Muci, K. R. Campos and D. A. Evans, J. Am. Chem. Soc., 1995, 117, 9075 CrossRef CAS.
- K. Nagata, S. Matsukawa and T. Imamoto, J. Org. Chem., 2000, 65, 4185 CrossRef CAS.
- Q. Xu, C. Q. Zhao and L. B. Han, J. Am. Chem. Soc., 2008, 130, 12648 CrossRef CAS.
- M. Stephan, B. Sterk, B. Modec and B. Mohar, J. Org. Chem., 2007, 72, 8010 CrossRef CAS.
- E. B. Kaloun, R. Merdès, J. P. Genêt, J. Uziel and S. Jugé, J. Organomet. Chem., 1997, 529, 455 CrossRef CAS.
- G. C. Lloyd-Jones and N. P. Taylor, Chem. – Eur. J., 2015, 21, 5423 CrossRef CAS.
- L. McKinstry and T. Livinghouse, Tetrahedron Lett., 1994, 35, 9319 CrossRef CAS.
- T. Imamoto, Chem. Rec., 2016, 16, 2659 CrossRef.
- K. M. M. Huihui, R. Shrestha and D. J. Weix, Org. Lett., 2017, 19, 340 CrossRef CAS.
- L. Horner, H. Winkler, A. Rapp, A. Mentrup, H. Hoffmann and P. Beck, Tetrahedron Lett., 1961, 2, 161 CrossRef.
- (a) O. Korpium and K. Mislow, J. Am. Chem. Soc., 1967, 89, 4784 CrossRef; (b) O. Korpium, R. A. Lewis, J. Chickos and K. Mislow, J. Am. Chem. Soc., 1968, 90, 4842 CrossRef.
- K. M. Pietrusiewicz and M. Zablocka, Chem. Rev., 1994, 94, 1375 CrossRef CAS.
- T. L. Emmick and R. L. Letsinger, J. Am. Chem. Soc., 1968, 90, 3459 CrossRef CAS.
- H. Y. Su and M. S. Taylor, J. Org. Chem., 2017, 82, 3173 CrossRef CAS.
- R. Bigler and A. Mezzetti, Org. Process Res. Dev., 2016, 20, 253 CrossRef CAS.
- D. Hérault, D. C. Nguyen, D. Nuel and G. Buono, Chem. Soc. Rev., 2015, 44, 2508 RSC.
- (a) G. X. Zhu, M. Terry and X. M. Zhang, Tetrahedron Lett., 1996, 37, 4475 CrossRef CAS; (b) G. X. Zhu, M. Terry and X. M. Zhang, J. Organomet. Chem., 1997, 547, 97 CrossRef CAS.
- B. Wolfe and T. Livinghouse, J. Am. Chem. Soc., 1998, 120, 5116 CrossRef CAS.
- I. Arenas, O. Boutureira, M. I. Matheu, Y. Diaz and S. Castillon, Eur. J. Org. Chem., 2015, 3666 CrossRef CAS.
- (a) T. Miura, H. Yamada, S. I. Kikuchi and T. Imamoto, J. Org. Chem., 2000, 65, 1877 CrossRef CAS; (b) Y. Zhu, Z. H. Yang, B. Q. Ding, D. L. Liu, Y. G. Liu, M. Sugiya, T. Imamoto and W. B. Zhang, Tetrahedron, 2015, 71, 6832 CrossRef; (c) Z. H. Yang, X. Wei, D. L. Liu, Y. G. Liu, M. Sugiya, T. Imamoto and W. B. Zhang, J. Organomet. Chem., 2015, 791, 41 CrossRef CAS; (d) Z. H. Yang, D. L. Liu, G. Liu, M. Sugiya, T. Imamoto and W. B. Zhang, Organometallics, 2015, 34, 1228 CrossRef CAS; (e) Z. H. Yang, C. Xia, D. L. Liu, Y. G. Liu, M. Sugiya, T. Imamoto and W. B. Zhang, Org. Biomol. Chem., 2015, 13, 2694 RSC.
- (a) B. S. Williams, P. Dani, M. Lutz, A. L. Spek and G. van Koten, Helv. Chim. Acta, 2001, 84, 3519 CrossRef CAS . See also: ; (b) D. Morales-Morales, R. E. Cramer and C. M. Jensen, J. Organomet. Chem., 2002, 654, 44 CrossRef CAS; (c) S. Medici, M. Gagliardo, S. B. Williams, P. A. Chase, S. Gladiali, M. Lutz, A. L. Spek, G. P. M. van Klink and G. van Koten, Helv. Chim. Acta, 2005, 88, 694 CrossRef CAS.
- B. Q. Ding, Z. F. Zhang, Y. Xu, Y. G. Liu, M. Sugiya, T. Imamoto and W. B. Zhang, Org. Lett., 2013, 15, 5476 CrossRef CAS.
- R. Huber, A. Passera and A. Mezzetti, Organometallics, 2018, 37, 396 CrossRef CAS.
- R. Huber, Iron meets stereogenic phosphorus: Fe(II) catalysts with chiral NPPN and PNP ligands, PhD thesis, ETH Zürich, 2018.
- R. Huber, A. Passera and A. Mezzetti, Adv. Synth. Catal., 2018, 360, 2990 CrossRef.
- T. Imamoto, M. Matsuo, T. Nonomura, K. Kishikawa and M. Yanagawa, Heteroat. Chem., 1993, 4, 475 CrossRef CAS.
- S. Lemouzy, D. H. Nguyen, V. Camy, M. Jean, D. Gatineau, L. Giordano, J. V. Naubron, N. Vanthuyne, D. Hérault and G. Buono, Chem. – Eur. J., 2015, 21, 15607 CrossRef CAS.
- W. M. Wang, L. J. Liu, C. Q. Zhao and L. B. Han, Eur. J. Org. Chem., 2015, 2342 CrossRef CAS.
- S. Sowa, M. Stankevič, A. Szmigielska, H. Małuszyńska, A. E. Kozioł and K. M. Pietrusiewicz, J. Org. Chem., 2015, 80, 1672 CrossRef CAS.
- (a) V. S. Chan, I. C. Stewart, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 2786 CrossRef CAS PubMed; (b) V. S. Chan, M. Chiu, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 6021 CrossRef CAS.
- (a) X. Z. Yang, Inorg. Chem., 2011, 50, 12836 CrossRef CAS PubMed; (b) G. R. Morello and K. H. Hopmann, ACS Catal., 2017, 7, 5847 CrossRef CAS.
- R. Langer, M. A. Iron, L. Konstantinovski, Y. Diskin-Posner, G. Leitus, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2012, 18, 7196 CrossRef CAS.
- J. I. Seeman, Chem. Rev., 1983, 83, 83 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |