
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a safely handleable synthetic equivalent of cyanonitrile oxide by 1,3-dipolar cycloaddition of nitroacetonitrile†
Nagatoshi
Nishiwaki
 *ab,
Yuta
Kumegawa
a,
Kento
Iwai
a and
Soichi
Yokoyama
ab
*ab,
Yuta
Kumegawa
a,
Kento
Iwai
a and
Soichi
Yokoyama
ab
aSchool of Environmental Science and Engineering, Kochi University of Technology, Tosayamada, Kami, Kochi 782-8502, Japan. E-mail: nishiwaki.nagatoshi@kochi-tech.ac.jp
bResearch Center for Material Science and Engineering, Kochi University of Technology, Tosayamada, Kami, Kochi 782-8502, Japan
First published on 4th June 2019
Abstract
Dianionic cyano-aci-nitroacetate affords 3-cyanoisoxazol(in)es upon heating with a range of dipolarophiles in the presence of hydrochloric acid. In this reaction, nitroacetonitrile is formed as an intermediate active species, which serves as a synthetic equivalent of cyanonitrile oxide that can participate in a 1,3-dipolar cycloaddition reaction.
The 1,3-dipolar cycloaddition of cyanonitrile oxide 1 (cyanofulminate or cyanogen N-oxide) is a useful preparative method for the synthesis of 3-cyanoisoxazol(in)es, which is superior to other methods such as direct substitution by a cyanide,1 chemical conversion of other functional groups,2 and ring construction3,4 because the five-membered ring can be formed in a single step. In addition, the products are readily transformed into polyfunctionalized compounds upon cleavage of the N–O bond. On the other hand, nitrile oxide 1 is too unstable to be isolated, although its generation can be confirmed by spectroscopy.5 Vacuum pyrolysis6 or base-induced dehydrochlorination7,8 of chloro(cyano)formoxime 2 are common methods used to generate nitrile oxide 1. In addition, the pyrolysis of dicyanofuroxan 39 and treating nitroacetonitrile 4 with ammonium cerium(III) nitrate10 are also effective approaches used to prepare 1 (Scheme 1). However, preparation of precursors 2 and 3 suffers from some drawbacks such as low yield, troublesome manipulations, and toxicity.11,12 In addition, nitroacetonitrile 4 should be handled as an explosive material (see ESI†).3,13 Hence, the development of a safe and concise method for generation of cyanonitrile oxide 1 is highly desired.
 | ||
| Scheme 1 Methods for the synthesis of cyanonitrile oxide 1. | ||
Meanwhile, we have studied the application of dianionic cyano-aci-nitroacetate 514 as a cyano(nitro)methylating agent in organic synthesis leading to the synthesis of functionalized heterocyclic systems.3,15 The structure of 5 can be regarded as a masked form of cyanonitrile oxide 1, based on which we envisaged that nitrile oxide 1 should be generated upon protonation of dianion 5 accompanied by decarboxylation and dehydration.
Although it is possible to isolate dianion 5, decarboxylation gradually occurs.14 Thus, 5 was generated in situ via a ring opening reaction of pyridinium salt 6 upon treatment with N-methylpyrrolidine. A solution of 6 and ethynylbenzene 7a in acetonitrile showed no change even when heated at 100 °C for 1 d in a sealed tube (Table 1, entry 1). On the other hand, a trace amount of 3-cyano-5-phenylisoxazole (8a)16 was obtained when the reaction was conducted under the same conditions in the presence of acetic acid (entry 2). The addition of a stronger acid such as p-toluenesulfonic acid and hydrochloric acid, was found to be effective (entries 3 and 4).
|
|
||
|---|---|---|
| Entry | Acid | Yield/% |
| 1 | — | 0 |
| 2 | AcOH | 5 |
| 3 | p-TsOH·H2O | 15 |
| 4 | HCl | 27 |
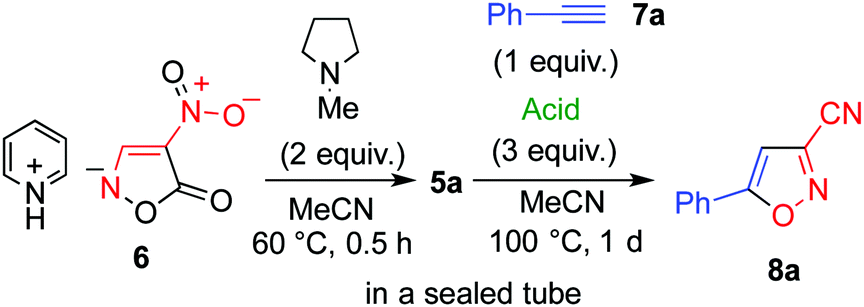
To obtain further insight into this reaction, each step was monitored using 1H NMR spectroscopy with deuterated acetonitrile used as the solvent (see ESI†). Cation exchange of 6 occurred after addition of 1 equivalent of N-methylpyrrolidine, and the ring opening reaction occurred leading to formation of the dianion, 5a, when another 1 equivalent of N-methylpyrrolidine was added. A new singlet signal appeared at 5.8 ppm when hydrochloric acid was added to the solution of 5a, in which nitroacetonitrile 4 or its anionic form 9a was formed as a result of the decarboxylation reaction, and either of these were considered to be the actual species in the cycloaddition reaction. Indeed, the formation of cycloadduct 8a was confirmed after heating this solution in the presence of 7a.
Subsequently, a more stable dipotassium salt, 5b, was employed from the viewpoint of its storage for a long period of time. Although 5a was more soluble in organic solvents, the reaction mixture was somewhat complicated because of the presence of pyridinium, pyrrolidinium, and ammonium chlorides, among which the last salt was formed during the acid-catalysed hydrolysis of acetonitrile. Thus, 5b was concluded to be more suitable for use as a reagent in organic synthesis.
Monopotassium salt 9b,14 derived from 5b, did not afford any cycloadduct 8a upon heating with ethynylbenzene 7a. However, the formation of 8a was confirmed when the same reaction was conducted in the presence of hydrochloric acid (Scheme 2). These results indicate that nitroacetonitrile 4 serves as a 1,3-dipole during the cycloaddition reaction.
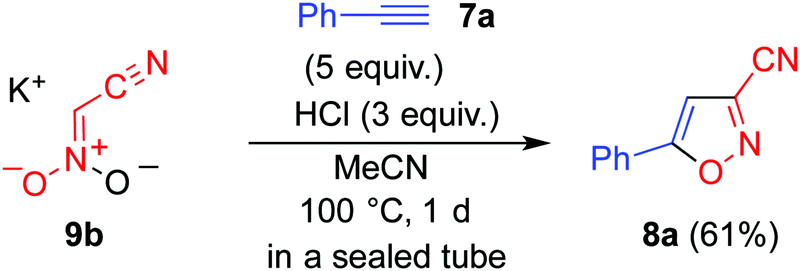 | ||
| Scheme 2 The cycloaddition reaction using monopotassium salt 9b under acidic conditions. | ||
The reaction conditions were optimized using dipotassium salt 5b (Table 2). A mixed solvent consisting of acetonitrile and water was suitable because it can dissolve both 5b and 7a (entries 1 and 2). However, no reaction occurred at lower temperature, which was presumably due to the low reactivity of nitroacetonitrile 4 when compared with conventional 1,3-dipoles such as nitrile oxide and nitrone (entries 2 and 3). Prolonging the reaction time was found to be effective for the reaction with cycloadduct 8a obtained in high yield after heating for 12 h (entries 4–6).‡
|
|
||||
|---|---|---|---|---|
| Entry | Solv. | Temp./°C | Time/h | Yield/% |
| 1 | MeCN | 100 | 24 | 78 |
| 2 | MeCN/H2O (1/4) | 100 | 24 | 90 |
| 3 | MeCN/H2O (1/4) | 70 | 24 | 0 |
| 4 | MeCN/H2O (1/4) | 100 | 3 | 0 |
| 5 | MeCN/H2O (1/4) | 100 | 6 | 43 |
| 6 | MeCN/H2O (1/4) | 100 | 12 | 96 |

Under the optimized conditions dipolarophiles 7 and 10 were subjected to the cycloaddition reaction (Scheme 3). Electron-deficient alkyne 7b underwent the cycloaddition reaction to afford 8b16 in quantitative yield. In contrast, alkenes 7c and 7d, which possess an electron-donating group furnished their corresponding cycloadducts 8c and 8d in moderate yields. Since considerable amounts of acetophenone derivatives were formed during these reactions, competitive hydration was considered to suppress the cycloaddition. trans-Stilbene 10e was also usable as a dipolarophile leading to the formation of diphenylisoxazoline 11e. Electron-deficient acrylates 10f and 10h also undergo the cycloaddition reaction efficiently, however, the ester functionality did not tolerate the reaction conditions, affording carboxylic acids 11g and 11i, respectively. Conversely, it was possible to use carboxylic acid 10i as a substrate without protection in this reaction, which afforded 11i in high yield.
 | ||
| Scheme 3 Synthesis of 3-cyanoisoxazol(in)es 8 and 11. | ||
This reaction was considered to proceed as shown in Scheme 4. Protonation of dianion 5 affords 12, which immediately undergoes decarboxylation to give nitroacetonitrile 4. The aci-nitro form of 4 serves as a 1,3-dipole and reacts with the dipolarophile to form a five-membered ring. Subsequent dehydration yields the final product. On the basis of this mechanism, the amount of acid necessary for this reaction is 2 equivalents. However, 3 equivalents of hydrochloric acid is necessary for an efficient reaction, which is presumably because the acid is also consumed by forming a salt with ammonia formed during the hydrolysis of acetonitrile.
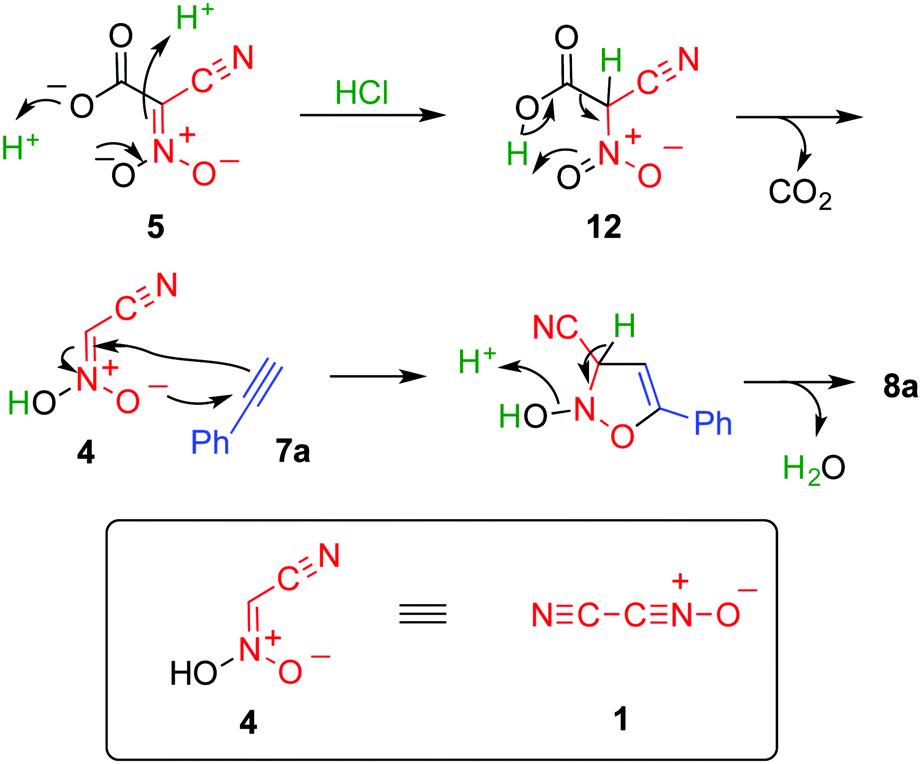 | ||
| Scheme 4 A plausible mechanism for our 1,3-dipolar cycloaddition reaction. | ||
To conclude, 3-cyanoisoxazol(in)es 8 and 11 were synthesized via the cycloaddition of nitroacetonitrile 4. While nitroacetonitrile 4 is an explosive material, dianion 5 is thermally stable and can be stored for a long period of time without any special care.3,13 Hence, dianion 5 serves as a safe handleable synthetic equivalent of cyanonitrile oxide 1, which can be used as a novel tool in organic synthesis.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. U. Dighe, S. Mukhopadhyay, S. Kolle, S. Kanojiya and S. Batra, Angew. Chem., Int. Ed., 2015, 54, 10926 CrossRef CAS PubMed; P. A. Wade and H. R. Hinney, J. Am. Chem. Soc., 1979, 101, 1319 CrossRef; P. A. Wade, J. Org. Chem., 1978, 43, 2020 CrossRef.
- Recent reports: N. A. Bumagin, A. V. Kletskov, S. K. Petkevich, I. A. Kolesnik, A. S. Lyakhov, L. S. Ivashkevich, A. V. Baranovsky, P. V. Kurman and V. I. Potkin, Tetrahedron, 2018, 74, 3578 CrossRef CAS; A. Nordqvist, G. O’Mahony, M. Friden-Saxin, M. Fredenwall, A. Hogner, K. L. Granberg, A. Aagaard, S. Baeckstroem, A. Gunnarsson, T. Kaminski, Y. Xue, A. Dellsen, E. Hansson, P. Hansson, I. Ivarsson, U. Karlsson, K. Bamberg, M. Hermansson, J. Georgsson, B. Lindmark and K. Edman, ChemMedChem, 2017, 12, 50 CrossRef PubMed; K. Natte, R. V. Jagadeesh, M. Sharif, H. Neumann and M. Beller, Org. Biomol. Chem., 2016, 14, 3356 RSC.
- K. Iwai, H. Asahara and N. Nishiwaki, J. Org. Chem., 2017, 82, 5409 CrossRef CAS PubMed.
- M. Tamura, T. Nishimura, N. Nishiwaki and M. Ariga, Heterocycles, 2004, 63, 1659 CrossRef CAS; N. Nishiwaki, T. Nogami, T. Kawamura, N. Asaka, Y. Tohda and M. Ariga, J. Org. Chem., 1999, 64, 6476 CrossRef; K. Gewald, P. Bellmann and H. J. Jaensch, Liebigs Ann. Chem., 1980, 1623 CrossRef.
- G. K. Williams and T. B. Brill, Combust. Flame, 1998, 114, 569 CrossRef CAS; R. Flammang, M. Barbieux-Flammang, P. Gerbaux, C. Wentrup and M. W. Wong, Bull. Soc. Chim. Belg., 1997, 106, 545 Search PubMed; T. Pasinski and N. P. C. Westwood, J. Phys. Chem., 1996, 100, 16856 CrossRef; T. Pasinski and N. P. C. Westwood, J. Chem. Soc., Chem. Commun., 1995, 1901 RSC.
- G. Maier and J. H. Teles, Angew. Chem., Int. Ed. Engl., 1987, 26, 155 CrossRef.
- A. P. Kozikowski and M. Adamczyk, J. Org. Chem., 1983, 48, 366 CrossRef CAS.
- J. S. Lee, Y. S. Cho, B. Y. Chung and A. N. Pae, Bioorg. Med. Chem., 2003, 13, 4117 CrossRef CAS; P. A. Wade and H. R. Hinney, Tetrahedron Lett., 1979, 139 CrossRef; C. Grundmann and H.-D. Frommeld, J. Org. Chem., 1966, 31, 4235 CrossRef.
- T. Shimizu, Y. Hayashi, T. Taniguchi and K. Teramura, Tetrahedron, 1985, 41, 727 CrossRef CAS.
- T. Sugiyama, Appl. Organomet. Chem., 1995, 9, 399 CrossRef CAS.
- H. Mohler, Protar, 1941, 7, 57 CAS.
- C. He and J. M. Shreeve, Angew. Chem., Int. Ed., 2016, 55, 772 CrossRef CAS PubMed; L. L. Fershtat, M. A. Epishina, A. S. Kulikov, I. V. Ovchinnikov, I. V. Ananyev and N. Nina, Tetrahedron, 2015, 71, 6764 CrossRef; T. M. Mel’nikova, T. S. Novikova, L. I. Khmel’nitskii and A. B. Sheremetev, Mendeleev Commun., 2001, 30 CrossRef.
- C. J. Thomas, Chem. Eng. News, 2009, 87(32), 4 CrossRef CAS.
- N. Nishiwaki, Y. Takada, Y. Inoue, Y. Tohda and M. Ariga, J. Heterocycl. Chem., 1995, 32, 473 CrossRef CAS.
- N. Nishiwaki, Curr. Med. Chem., 2017, 24, 3728 CrossRef CAS PubMed; H. Asahara, K. Muto and N. Nishiwaki, Tetrahedron, 2014, 70, 6522 CrossRef; N. Nishiwaki, T. Nogami, T. Kawamura, N. Asaka, Y. Tohda and M. Ariga, J. Org. Chem., 1999, 64, 6476 CrossRef; N. Nishiwaki, T. Nogami, C. Tanaka, F. Nakashima, Y. Inoue, N. Asaka, Y. Tohda and M. Ariga, J. Org. Chem., 1999, 64, 2160 CrossRef PubMed.
- N. A. Bumagin, A. V. Kletskov, S. K. Petkevich, I. A. Kolesnik, A. S. Lyakhov, L. S. Ivashkevich, A. V. Baranovsky, P. V. Kurman and V. I. Potkin, Chem. Heterocycl. Compd., 2014, 49, 1515 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods, monitoring experiments using 1H NMR, DSC measurement, and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c9cc03875b |
| ‡ Typical procedure: in a screw capped test tube, dipotassium salt 5b (41.2 mg, 0.2 mmol) was dissolved into a mixed solvent of MeCN/H2O (v/v = 1/1, 2 mL). After adding 1-ethynyl-4-trifluoromethylbenzene 7b (144 μL, 1 mmol) and 1 M HCl (3 mL, 3 mmol), the resultant mixture was heated at 100 °C for 12 h in a sealed tube. The solvent was removed under reduced pressure, and MeCN (10 mL) was added to the residue. After filtration to remove the insoluble material, the filtrate was concentrated to afford 3-cyano-5-(4-trifluoromethylphenyl)isoxazole 8b (239 mg, 1 mmol, quant.) as a pale yellow solid. M.p. 92–95 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.04 (s, 1H), 8.05 (d, J = 8.2 Hz, 2H), 8.23 (d, J = 8.2 8.2 Hz, 2H); 13C NMR (100 MHz, DMSO-d6) δ 105.0 (CH), 110.2 (C), 123.7 (C, q, J = 271 Hz), 126.4 (CH, d, J = 3.5 Hz), 126.9 (CH), 128.7 (C), 131.3 (C, q, J = 32.1 Hz), 140.3 (C), 170.3 (C); IR (ATR/cm−1) 2261, 1570, 1400, 1319; HRMS (ESI/TOF) calcd for (M + H+) C11H6F3N2O: 239.0427, found: 239.0428. |
| This journal is © The Royal Society of Chemistry 2019 |