
Transition-metal- and oxidant-free directed anodic C–H sulfonylation of N,N-disubstituted anilines with sulfinates†
Yan-Chen
Wu
a,
Shuai-Shuai
Jiang
a,
Shu-Zheng
Luo
a,
Ren-Jie
Song
*a and
Jin-Heng
Li
 *ab
*ab
aKey Laboratory of Jiangxi Province for Persistent Pollutants Control and Resources Recycle, Nanchang Hangkong University, Nanchang 330063, China. E-mail: srj0731@hnu.edu.cn
bState Key Laboratory of Chemo/Biosensing and Chemometrics, Hunan University, Changsha 410082, China. E-mail: jhli@hnu.edu.cn
First published on 28th June 2019
Abstract
A new, practical directed anodic C–H sulfonylation of N,N-disubstituted anilines with sodium sulfinates for producing o- or p-amino arylsulfones and diarylsulfones is described. Employing the anodic strategy, the reaction proceeds efficiently under mild (room temperature) and transition-metal- and chemical oxidant-free conditions, and enables the formation of C–S bonds via directed activation of ortho- or para-C–H bond to the amino group with broad substrate scope and excellent site selectivity.
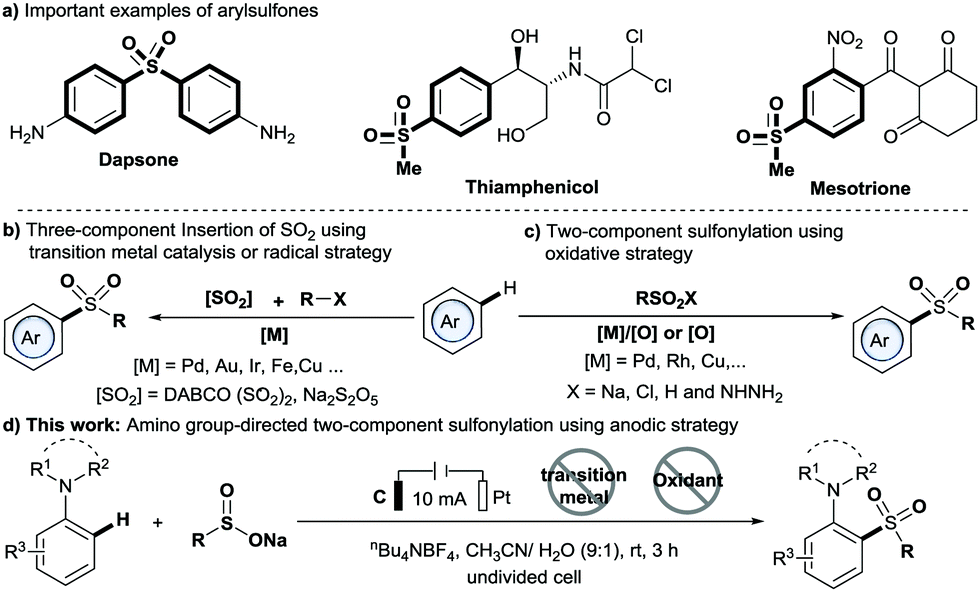
Arylsulfones are recognized as an important class of organic molecule because they have unique biological activity and wide applications ranging from chemical to physical.1,2 For example, dapsone is a preferred drug for leprosy (Scheme 1a).3 Traditionally, methods for the synthesis of arylsulfones generally proceed by means of Friedel–Crafts-type sulfonation or oxidation of sulfides.4,5 In recent years, C–H sulfonylation reactions using either the C–H activation or the radical strategy have been developed (Scheme 1b and c).6–10 Particularly, the directed C–H activation strategy has attracted much attention due to its high atom and step economies as well as excellent site selectivity, which has been widely applied in C–H sulfur dioxide insertion reactions (Scheme 1b) and oxidative C–H sulfonylation (Scheme 1c). However, most of these methods require expensive noble transition metal catalysts, stoichiometric amounts of oxidants and/or relatively high temperature. Thus, the development of new, efficient strategies toward arylsulfone derivatives under mild, metal-free and chemical oxidant-free conditions is appealing.
 | ||
| Scheme 1 Important sulfones and C–H sulfonylation. | ||
Electrochemical synthesis has become a powerful tool in organic synthesis due to its safety and sustainability.11 Importantly, the electrochemical strategy has been applied to C–H functionalization, albeit with the majority involving the use of transition metal catalysis. In 2018, Lei and co-workers reported an electrochemical direct oxidative C–H sulfonylation of (hetero)arenes using sulfonyl hydrazides as sulfonylating agents.12 Very recently, Waldvogel and co-workers reported a novel metal- and reagent-free electrochemical strategy for the synthesis of arylsulfones by direct sulfonylation of phenols with sodium sulfinates.13 However, the method is limited by the requirement of special highly substituted phenols to achieve excellent regioselectivity. To our knowledge, methods for directed sulfonylation of anilines using the electrochemical method have never been reported. Herein, we report a new anodic C–H sulfonylation of N,N-disubstituted anilines with sodium sulfinates for producing o- or p-amino arylsulfones and diarylsulfones (Scheme 1d). The method enables the regiospecific construction of the C–S bond under mild and transition-metal- and chemical oxidant-free conditions, and provides a practical and sustainable tool to access the valuable C,S-coupling products from common reaction components: N,N-disubstituted anilines and sodium sulfinates.
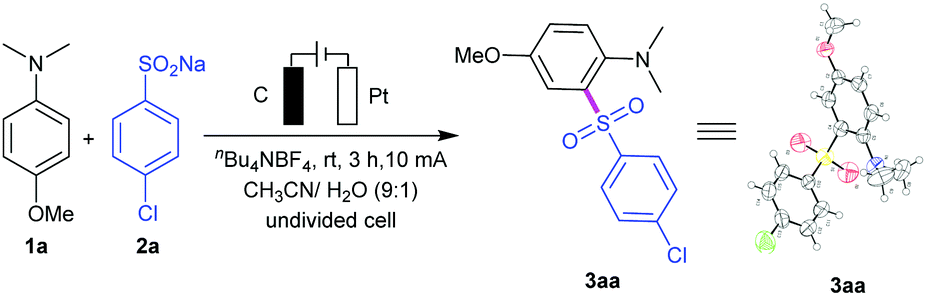
By utilizing nBu4NBF4 as the electrolyte and CH3CN/H2O as co-solvents, the directed anodic C–H sulfonylation between 4-methoxy-N,N-dimethylaniline (1a) and sodium 4-chlorobenzenesulfinate (2a) was performed, affording the desired C,S-coupling product 3aa in a 91% yield under 10 mA constant current for 3 h in an undivided cell (a three-necked round-bottomed flask) equipped with a carbon rod anode and a platinum plate cathode (entry 1; Table 1). Changing the operating current of this transformation resulted in a decrease in yield (entries 2 and 3). Control experiments showed that the reaction had less reactivity when using nBu4NBr or LiClO4 as the electrolyte (entries 4 and 5). The yield decreased sharply to 67% using MeOH/H2O instead of CH3CN/H2O (entry 6). We found that H2O was necessary to increase the solubility of the salts (entry 7). However, use of water alone as the medium led to diminishing yield (entry 8). Two other electrode material combinations were screened, which proved use of graphite rod as the anode and platinum plate as the cathode to be the best choice (entry 1 versus entries 9, 10). The method showed no apparent sensitivity to oxygen and therefore could be performed under air atmosphere conditions (entry 11). The reaction could not take place under no electric current condition (entry 12). Notably, the synthetic potential of this reaction was then evaluated by performing a 6.6 mmol (1 g) scale reaction, giving 1.64 g of 3aa (71% yield) after 18 h (entry 13). The reaction could also give 3aa in 27% yield under Waldvogel's condition, and give by-product 2,6-bis((4-chlorophenyl)sulfonyl)-4-methoxy-N,N-dimethylaniline (4aa) in 12% yield.
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yield (%) |
| a Reaction conditions: carbon rod anode, platinum plate cathode, constant current = 10 mA, 1a (0.3 mmol), 2a (4 equiv.), nBu4NBF4 (0.1 mmol), CH3CN/H2O (9.0 mL/1.0 mL), room temperature under air atmosphere for 3 h. b 1a (1 g, 6.6 mmol) for 18 h, 13% of 1a was recovered. c By-product 2,6-bis((4-chlorophenyl)sulfonyl)-4-methoxy-N,N-dimethylaniline (4aa) was isolated in 12% yield. | ||
| 1 | None | 91 |
| 2 | 5 mA instead of 10 mA, 6 h | 90 |
| 3 | 15 mA instead of 10 mA, 1.5 h | 83 |
| 4 | n Bu4NBr instead of nBu4NBF4 | 85 |
| 5 | LiClO4 instead of nBu4NBF4 | 62 |
| 6 | CH3OH/H2O (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) instead of MeCN/H2O (9:1) :1) instead of MeCN/H2O (9:1) |
67 |
| 7 | Without H2O | Trace |
| 8 | Without MeCN | 26 |
| 9 | C(+)|C(−) instead of C(+)∣Pt(−) | 75 |
| 10 | Pt(+)|C(−) instead of C(+)∣Pt(−) | 85 |
| 11 | Under Ar | 88 |
| 12 | No electric current | 0 |
| 13b | None | 71 |
| 14c | No nBu4NBF4, HFIP:H2O (9:1) as solvent |
27 |
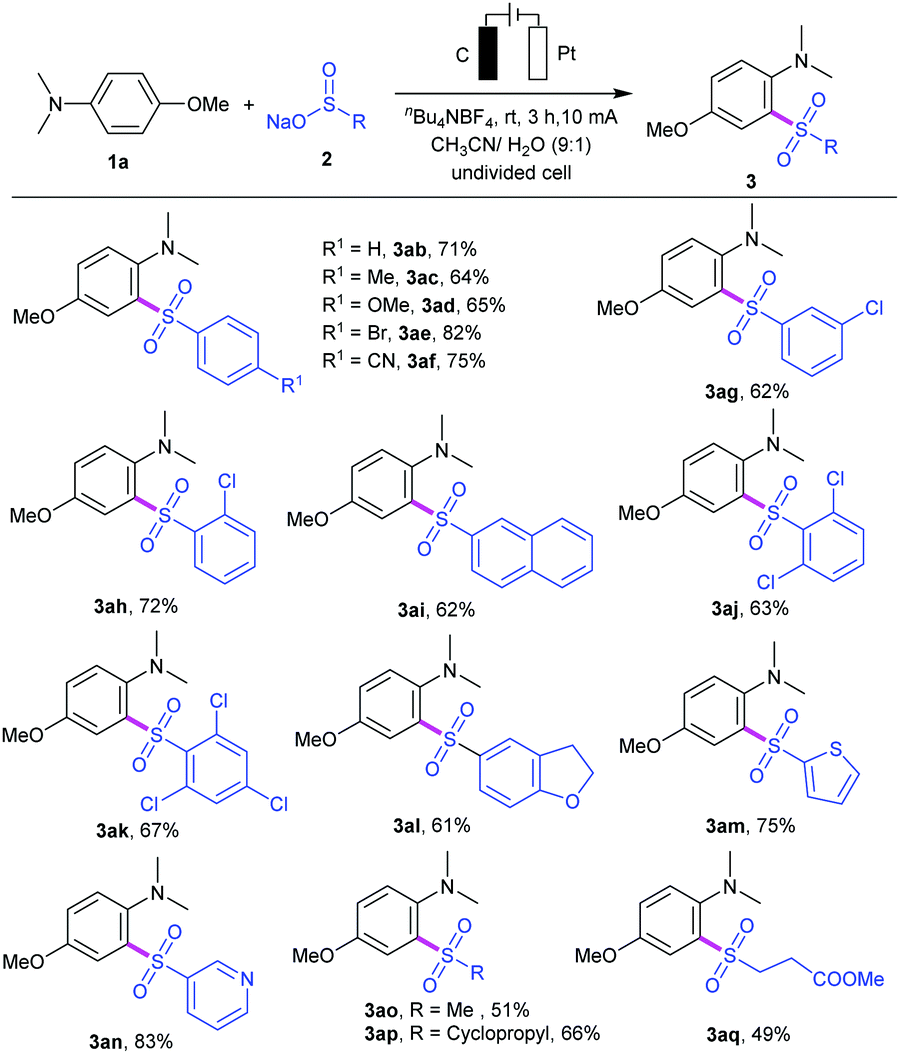
We then investigated the substrate scope of the reaction by varying the substituents of the aryl and alkyl sulfinates, and the results are summarized in Table 2. Sulfinate derivatives 2b–f with different functional groups, such as electron-donating groups (R1 = Me, OMe) and electron-withdrawing groups (R1 = Br, CN) on the para-site of aryl ring, were suitable for this electrooxidative C–H sulfonylation and furnished the desired sulfonyl aniline in moderate to excellent yields (3ab–af). The position of the substituents on the aryl ring had a minor effect on the efficiency of this transformation. For instance, meta- or ortho-substituted sulfinates 2g and 2h proceeded smoothly to give the corresponding products 3ag and 3ah in 62% and 72% yields, respectively. To our delight, sodium naphthalene-2-sulfinate 2i was compatible with the current conditions and gave the corresponding sulfonyl aniline 3ai in 62% yield. Subsequently, we also examined the reactivity of di- or trisubstituted sulfinates 2j–k, and the results showed that they could be successfully reacted with aniline 1a (products 3aj and 3ak). Notably, 2,3-dihydrobenzofuranyl, heterocyclic thienyl and 3-pyridyl sulfinates could work well in the reaction to provide the corresponding heterocyclic products 3al–3an in 61–83% yields. Moreover, alkyl sulfinates, including sodium methanesulfinate 2n, sodium 3-methoxy-3-oxopropane-1-sulfinate 2o and sodium cyclopropanesulfinate 2p, could be applied under this condition, providing the corresponding sulfones 3ao–aq in moderate yields.
| a Reaction conditions: carbon rod anode, platinum plate cathode, constant current = 10 mA, 1a (0.3 mmol), 2a (4 equiv.), nBu4NBF4 (0.1 mmol), CH3CN/H2O (9.0 mL/1.0 mL), room temperature under air atmosphere for 3 h. |
|---|
|
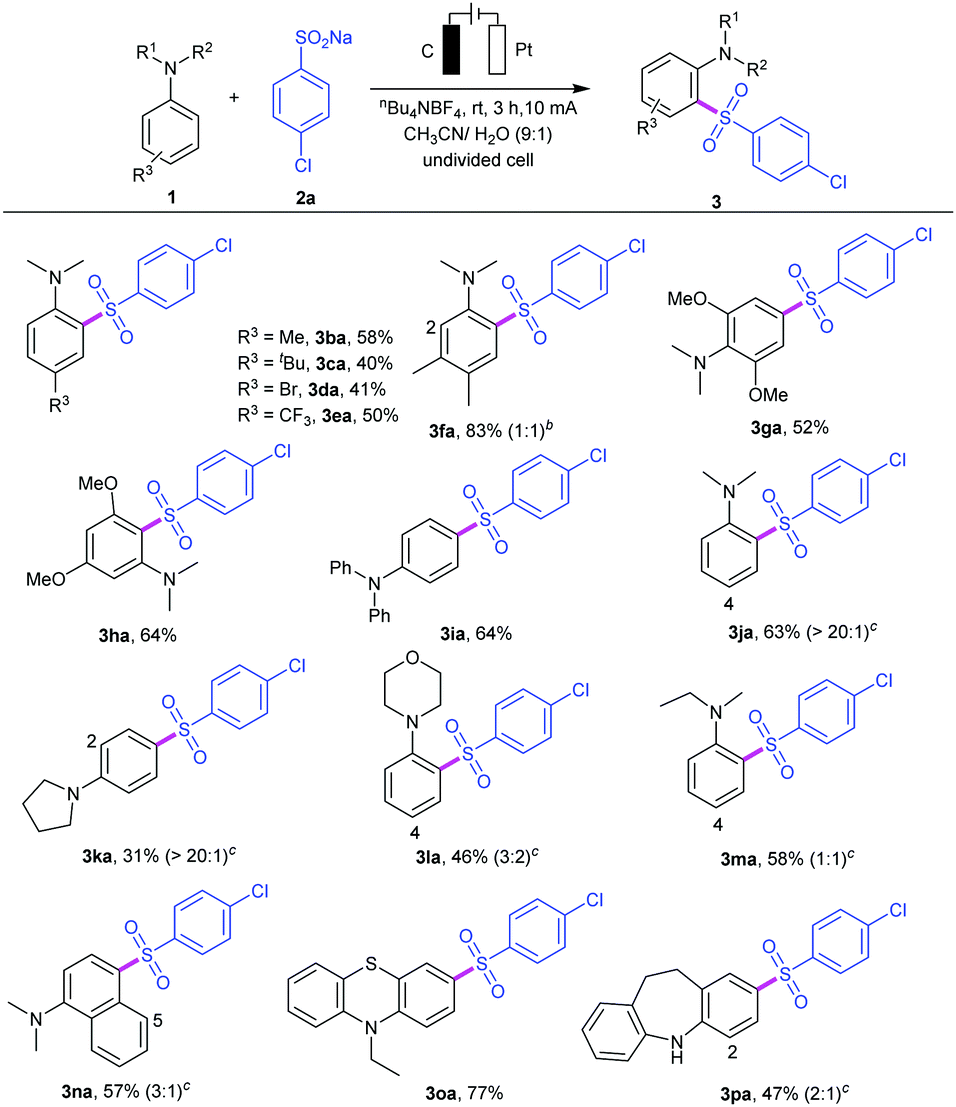
To examine the synthetic utility of this electrooxidative arylsulfonylation protocol, the scope of N,N-disubstituted anilines 1 was also investigated (Table 3). A series of N,N-disubstituted anilines can be tolerated regardless of the electronic and steric effects. For example, para-substituted anilines 1b–e bearing both electron-donating groups (R3 = Me, tert-butyl) and electron-withdrawing groups (R3 = Br, CF3) led to sulfonyl anilines 3ba–3ea in 40–58% yields. Asymmetric disubstituted aniline 1f could react with sodium 4-chlorobenzenesulfinate 2a, giving a mixture of products 3fa in 83% yield. Moreover, symmetrical disubstituted anilines 1g and 1h were also amenable to this protocol, affording the reasonable products 3ga and 3ha in 52% and 64% yields, respectively. A survey of aniline derivatives with different aromatic groups and alkyl groups substituted at the nitrogen atom showed reactivity with sulfinate 2a. In addition, N,N-dialkyl-substituted anilines, such as N,N-dimethylaniline 1j, N-phenyl heterocyclic amines 1k–l, and N-ethyl-N-methylaniline 1m, were also tolerated but led to lower yields. It was observed that, in most cases, this electrooxidative sulfonylation reaction occurred predictably at the ortho- and para-positions with respect to the amino substituent. The ortho- and para-amino arylsulfones were obtained at the same time, and the site selectivity may be controlled by the steric hindrance effect. Notably, N,N-dimethylnaphthalen-1-amine 1n was also suitable for this transformation (product 3na).14 As we expected, C4,C5-sulfonated products can be separated under the same catalytic system. This methodology could also be applied to some drug structure derivatives. For example, the reaction of 10-ethyl-10H-phenothiazine (1o) gave the desired product 3oa in 67% yield, and iminodibenzyl (1p) also showed good reactivity, affording the C2- and C4-sulfonated product 3pa in 47% yield under the same reactions. Phenothiazine derivatives have been reported to be a class of antipsychotics and iminodibenzyl is an important class of pharmaceutical intermediates for synthesis of specific analgesic and antipsychotic agents.
| a Reaction conditions: carbon rod anode, platinum plate cathode, constant current = 10 mA, 1a (0.3 mmol), 2a (4 equiv.), nBu4NBF4 (0.1 mmol), CH3CN/H2O (9.0 mL/1.0 mL), room temperature under atmosphere for 3 h. b This compound is obtained as a mixture of regioisomers that could not be separated by standard chromatography. c This compound could be separated by standard chromatography; for details, see ESI. |
|---|
|
Based on the experiments, CV results (see ESI†) and previous reports,15,16 a possible mechanism for the electrochemical oxidative direct sulfonylation of anilines is proposed in Scheme 2. Firstly, sodium 4-chlorobenzenesulfinate 2a is oxidized at the anode to give the oxygen-centered radical I, and the oxygen-centered radical resonates to the more stable sulfonyl radical II. At the same time, 4-methoxy-N,N-dimethylaniline 1a might also be oxidized by the carbon anode to generate a radical cation III. C–S bond was likely to be formed from the radical/radical cross-coupling between radical cation III and sulfonyl radical II, and finally resulted in the desired product 3aa after a deprotonation process. At the cathode, the co-solvent H2O could be reduced to give hydrogen gas during the reaction.
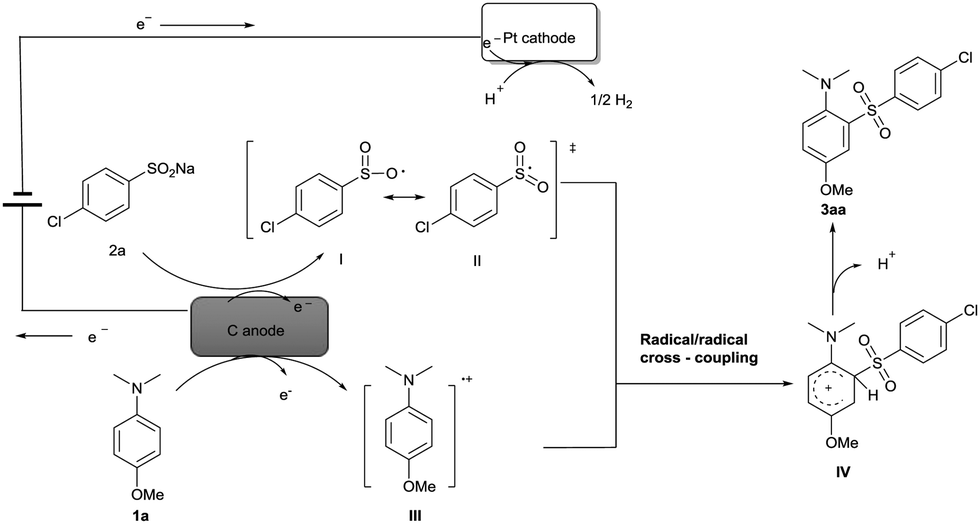 | ||
| Scheme 2 Possible reaction mechanism. | ||
In conclusion, we have established a general protocol to couple N,N-disubstituted anilines with aryl, heteroaryl and alkyl sulfinates in the absence of any transition metal catalyst promoted by electrochemical means at room temperature using undivided electrochemical cells. A broad range of aryl and heteroaryl sulfones could be constructed under this catalyst system and this method tolerated many functional groups. Importantly, the reaction conditions were compatible with some drug structure derivatives. Further efforts to understand the electrochemical oxidative reaction mechanism are currently underway in our laboratory.
We thank the Natural Science Foundation of China (no. 51878326, 21762030, 21625203 and 21871126) and Jiangxi Province Science and Technology Project (no. 20171BCB23055 and 20181BAB213002) for financial support.
Conflicts of interest
The authors declare no conflict of interest.Notes and references
- (a) Q. A. Acton, Sulfones-Advances in Research and Application, Scholarly Editions, Atlanta, 2013 Search PubMed; (b) S. Patai, Z. Rappoport and C. J. M. Stirling, The Chemistry of Sulphones and Sulphoxides, Wiley, New York, 1988 CrossRef; (c) J.-J. Li, Name Reactions in Heterocyclic Chemistry, John Wiley & Sons, Hoboken, 2005 Search PubMed; (d) T. Gilligan and W. Oh, Prostate, 2001, 3, 42 CrossRef.
- (a) N.-W. Liu, S. Liang and G. Manolikakes, Synthesis, 2016, 1939 CAS; (b) S. Shaaban, S. Liang, N.-W. Liu and G. Manolikakes, Org. Biomol. Chem., 2017, 15, 1947 RSC; (c) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291 RSC; (d) Y. Fang, Z. Luo and X. Xu, RSC Adv., 2016, 6, 59661 RSC; (e) D. C. Meadows, T. Sanchez, N. Neamati, T. W. North and J. Gervay-Hague, Bioorg. Med. Chem., 2007, 15, 1127 CrossRef CAS.
- (a) J. L. Fernandes Vieira, J. G. Bardarez Riveira, A. d. N. Silva Martins, J. P. da Silva and C. G. Salgado, Braz. J. Infect. Dis., 2010, 14, 319 CrossRef; (b) A. Chakraborty, A. K. Panda, R. Ghosh and A. Biswas, Arch. Biochem. Biophys., 2019, 665, 107 CrossRef CAS.
- For the Friedel–Crafts type sulfonation see: (a) G. A. Olah, T. Mathew and G. K. Surya Prakash, Chem. Commun., 2001, 1696 RSC; (b) M. Peyronneau, M.-T. Boisdon, N. Roques, S. Mazières and C. Le Roux, Eur. J. Org. Chem., 2004, 4636 CrossRef CAS; (c) K. Smith, G. M. Ewart, G. A. El-Hiti and K. R. Randles, Org. Biomol. Chem., 2004, 2, 3150 RSC; (d) N.-W. Liu, S. Liang and G. Manolikakes, Synthesis, 2016, 1939 CAS.
- For the oxidation of sulphide see: (a) B. Maleki, S. Hemmati, A. Sedrpoushan, S. S. Ashrafi and H. Veisi, RSC Adv., 2014, 4, 40505 RSC; (b) J. J. Boruah, S. P. Das, S. R. Ankireddy, S. R. Gogoi and N. S. Islam, Green Chem., 2013, 15, 2944 RSC.
- For the ortho C–H activation see: (a) X. Tang, L. Huang, Y. Xu, J. Yang, W. Wu and H. Jiang, Angew. Chem., Int. Ed., 2014, 53, 4205 CrossRef CAS; (b) Z. Wu, H. Song, X. Cui, C. Pi, W. Du and Y. Wu, Org. Lett., 2013, 15, 1270 CrossRef CAS; (c) S. Liang, N.-W. Liu and G. Manolikakes, Adv. Synth. Catal., 2016, 358, 159 CrossRef CAS; (d) O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298 CrossRef CAS; (e) F. Xiao, S. Chen, Y. Chen, H. Huang and G.-J. Deng, Chem. Commun., 2015, 51, 652 RSC; (f) W.-H. Rao and B.-F. Shi, Org. Lett., 2015, 17, 2784 CrossRef CAS.
- For the insertion of sulfur dioxide see (a) G. Qiu, K. Zhou and J. Wu, Chem. Commun., 2018, 54, 12561 RSC; (b) G. Qiu, L. Lai, J. Cheng and J. Wu, Chem. Commun., 2018, 54, 10405 RSC; (c) A. S. Deeming, C. J. Russell, A. J. Hennessy and M. C. Willis, Org. Lett., 2014, 16, 150 CrossRef CAS; (d) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2014, 53, 10204 CrossRef CAS; (e) D. Zheng, J. Yu and J. Wu, Angew. Chem., Int. Ed., 2016, 55, 11925 CrossRef CAS; (f) K. Zhou, J. Zhang, L. Lai, J. Cheng, J. Sun and J. Wu, Chem. Commun., 2018, 54, 7459 RSC; (g) M. Wang, B.-C. Tang, J.-G. Wang, J.-C. Xiang, A.-Y. Guan, P.-P. Huang, W.-Y. Guo, Y.-D. Wu and A.-X. Wu, Chem. Commun., 2018, 54, 7641 RSC; (h) X. Gong, J. Chen, L. Lai, J. Cheng, J. Sun and J. Wu, Chem. Commun., 2018, 54, 11172 RSC.
- For the oxidative C–H/X–H cross coupling see (a) Y. Zhao, H. Wang, X. Hou, Y. Hu, A. Lei, H. Zhang and L. Zhu, J. Am. Chem. Soc., 2006, 128, 15048 CrossRef CAS; (b) L.-H. Lu, S.-J. Zhou, W.-B. He, W. Xia, P. Chen, X. Yu, X. Xu and W.-M. He, Org. Biomol. Chem., 2018, 16, 9064 RSC; (c) H. Yi, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016 CrossRef CAS; (d) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787 CrossRef CAS; (e) L.-Y. Xie, S. Peng, F. Liu, J.-Y. Yi, M. Wang, Z. Tang, X. Xu and W.-M. He, Adv. Synth. Catal., 2018, 360, 4259 CrossRef CAS; (f) L.-Y. Xie, J. Qu, S. Peng, K.-J. Liu, Z. Wang, M.-H. Ding, Y. Wang, Z. Cao and W.-M. He, Green Chem., 2018, 20, 760 RSC.
- (a) W. Ge and Y. Wei, Green Chem., 2012, 14, 2066 RSC; (b) C. Wu, L.-H. Lu, A.-Z. Peng, G.-K. Jia, C. Peng, Z. Cao, Z. Tang, W.-M. He and X. Xu, Green Chem., 2018, 20, 3683 RSC; (c) C. D. Prasad, S. J. Balkrishna, A. Kumar, B. S. Bhakuni, K. Shrimali, S. Biswas and S. Kumar, J. Org. Chem., 2013, 78, 1434 CrossRef CAS; (d) W.-J. Hao, Y. Du, D. Wang, B. Jiang, Q. Gao, S.-J. Tu and G. Li, Org. Lett., 2016, 18, 1884 CrossRef CAS; (e) P. Katrun, C. Mueangkaew, M. Pohmakotr, V. Reutrakul, T. Jaipetch, D. Soorukram and C. Kuhakarn, J. Org. Chem., 2014, 79, 1778 CrossRef CAS; (f) L.-Y. Xie, S. Peng, J.-X. Tan, R.-X. Sun, X. Yu, N.-N. Dai, Z.-L. Tang, X. Xu and W.-M. He, ACS Sustainable Chem. Eng., 2018, 6, 16976 CrossRef CAS; (g) L.-Y. Xie, Y.-J. Li, J. Qu, Y. Duan, J. Hu, K.-J. Liu, Z. Cao and W.-M. He, Green Chem., 2017, 19, 5642 RSC; (h) H. Yue, C. Zhu and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 1371 CrossRef CAS; (i) L.-Y. Xie, S. Peng, F. Liu, G.-R. Chen, W. Xia, X. Yu, W.-F. Li, Z. Cao and W.-M. He, Org. Chem. Front., 2018, 5, 2604 RSC.
- T. C. Johnson, Bryony L. Elbert, A. J. M. Farley, T. W. Gorman, C. Genicot, B. Lallemand, P. Pasau, J. Flasz, J. L. Castro, M. MacCoss, D. J. Dixon, R. S. Paton, C. J. Schofield, M. D. Smith and M. C. Willis, Chem. Sci., 2018, 9, 629 RSC.
- (a) Y.-Y. Jiang, K. Xu and C.-C. Zeng, Chem. Rev., 2018, 118, 4485 CrossRef CAS; (b) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706 CrossRef CAS; (c) Y. Zhao and W. Xia, Chem. Soc. Rev., 2018, 47, 2591 RSC; (d) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786 RSC; (e) N. Sauermann, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Catal., 2018, 8, 7086 CrossRef CAS; (f) X. Gao, P. Wang, L. Zeng, S. Tang and A. Lei, J. Am. Chem. Soc., 2018, 140, 4195 CrossRef CAS; (g) H.-B. Zhao, P. Xu, J. Song and H.-C. Xu, Angew. Chem., Int. Ed., 2018, 57, 15153 CrossRef CAS; (h) M.-J. Luo, B. Liu, Y. Li, M. Hu and J.-H. Li, Adv. Synth. Catal., 2019, 361, 1538 CrossRef CAS.
- Y. Yuan, Y. Yu, J. Qiao, P. Liu, B. Yu, W. Zhang, H. Liu, M. He, Z. Huang and A. Lei, Chem. Commun., 2018, 54, 11471 RSC.
- J. Nikl, S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Chem. – Eur. J., 2019, 25, 6891 CrossRef CAS.
- CCDC 1903203 (3aa) 1923102 (3na) contain the supplementary crystallographic data for this paper†.
- (a) P. Wang, S. Tang, P. Huang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 3009 CrossRef CAS; (b) M.-L. Feng, L.-Y. Xi, S.-Y. Chen and X.-Q. Yu, Eur. J. Org. Chem., 2017, 2746 CrossRef CAS; (c) Y.-Y. Jiang, S. Liang, C.-C. Zeng, L.-M. Hu and B.-G. Sun, Green Chem., 2016, 18, 6311 RSC; (d) Y. Yuan, Y. Cao, Y. Lin, Y. Li, Z. Huang and A. Lei, ACS Catal., 2018, 8, 10871 CrossRef CAS; (e) K. Liu, S. Tang, T. Wu, S. Wang, M. Zou, H. Cong and A. Lei, Nat. Commun., 2019, 10, 639 CrossRef.
- (a) Y. Gao, H. Mei, J. Han and Y. Pan, Chem. – Eur. J., 2018, 24, 17205 CrossRef CAS; (b) M.-W. Zheng, X. Yuan, Y.-S. Cui, J.-K. Qiu, G. Li and K. Guo, Org. Lett., 2018, 20, 7784 CrossRef CAS; (c) X.-Y. Qian, S.-Q. Li, J. Song and H.-C. Xu, ACS Catal., 2017, 7, 2730 CrossRef CAS; (d) C. Zhang, Y. Chen and G. Yuan, Chin. J. Chem., 2016, 34, 1277 CrossRef CAS; (e) P. Huang, P. Wang, S. Tang, Z. Fu and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 8115 CrossRef CAS PubMed; (f) D. Liu, H.-X. Ma, P. Fang and T.-S. Mei, Angew. Chem., Int. Ed., 2019, 58, 5033 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1903203 (3aa) 1923102 (3na). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc03789f |
| This journal is © The Royal Society of Chemistry 2019 |