
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aerosol-based synthesis of pure and stable amorphous calcium carbonate†
Jacinta M.
Xto
 *ab,
Camelia N.
Borca
a,
Jeroen A.
van Bokhoven
ab and
Thomas
Huthwelker
*a
*ab,
Camelia N.
Borca
a,
Jeroen A.
van Bokhoven
ab and
Thomas
Huthwelker
*a
aPaul Scherrer Institut, 5232 Villigen, Switzerland. E-mail: Thomas.huthwelker@psi.ch
bInstitute for Chemical and Bioengineering, ETH Zürich, 8093 Zürich, Switzerland
First published on 20th August 2019
Abstract
A facile aerosol-based method for the synthesis of pure and stable amorphous calcium carbonate (ACC) is presented. The method relies on the instantaneous carbonation of calcium hydroxide aerosols with carbon dioxide followed by rapid drying of the freshly formed ACC. The ACC display extended stability against humidity induced crystallization.
The polymorphism of calcium carbonate is extensively studied in chemistry, mineralogy and earth science and most recently more intensively for carbon dioxide capture and storage technologies.1,2 Calcite, aragonite and vaterite are anhydrous polymorphs of CaCO3 and ikaite and monohydrocalcite are hydrated polymorphs.3,4 During precipitation reactions of calcium carbonate from supersaturated solutions, formation of transient amorphous calcium carbonate (ACC) has been reported in a variety of synthetic systems.5–13 In calcifying organisms, transient ACC formation has also been extensively reported10,14 and it has been postulated that the structure of the ACC largely determines which crystalline phase is formed.15,16 The remarkable architectures displayed in the brittle star lens, nacre and the sea urchin spicules, for example, have been associated to the remarkable control over ACC crystallization.10,17–19
With increasing evidence pointing to the role of ACC in formation of biologically relevant functional materials, there has been an increase of interest in ACC formation and crystallization for their potential application in industry and materials synthesis.20–24 A variety of ACC synthesis methods have been developed, most of which rely on additives such as magnesium/sulphate ions or organic additives/solvents.6,25–27 The presence of these additives affects the nature and crystallization behaviour of ACC in different ways. The synthesis protocol, presence of co-precipitated ions, amount of mobile water, temperature, pH and drying conditions are among the factors that have been suggested to affect ACC crystallization.16,28–33 However, without a pure ACC reference, it is not possible to correlate these factors. Therefore, to fully understand and exploit the potential of ACC, there is a need to develop facile and reproducible synthesis methods of additive-free and stable ACC reference.
In this work, we present a facile and reproducible aerosol-based method for the formation of pure and stable ACC. The method relies on the rapid reaction of calcium hydroxide aerosols with carbon dioxide (eqn (1)). The subsequent rapid drying of the ACC aerosols using a diffusion drier prevents crystallization and results in the formation of pure, stable and dry ACC.
| Ca(OH)2(aq) + CO2(g) → CaCO3(s) + H2O(l) | (1) |
Fig. 1 shows a schematic of the aerosol-based system, consisting of a commercial available atomizer model 3076 from TSI,34 a diffusion drier and a syringe pump (SPL with touch screen). A high velocity gas jet forms from the expansion of 2.5 bars carbon dioxide through a 0.0135 mm orifice into the atomizer. The carbon dioxide jet atomizes a fresh calcium hydroxide solution, which is continuously fed with a syringe pump at a rate of 1 ml min−1 into a vertical passage.
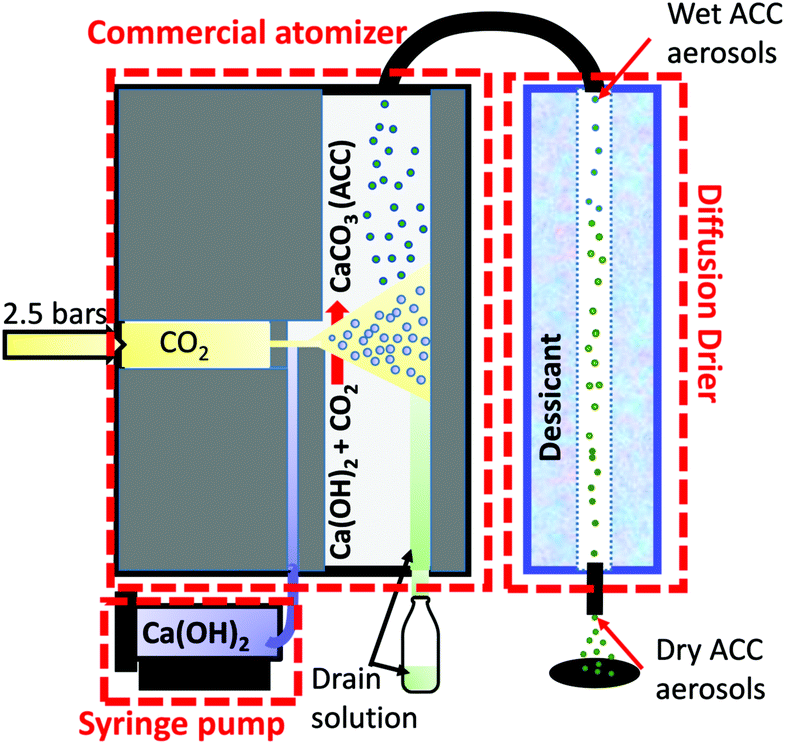 | ||
| Fig. 1 Schematic of the aerosol synthesis of dry amorphous calcium carbonate (ACC). | ||
Calcium hydroxide, being very basic (pH ∼ 12), rapidly reacts with the carbon dioxide to form ACC aerosols. A fine spray consisting of droplets of about 300 nm34 in size follows the gas flow into a gas permeable tube in the diffusion dryer. Through impaction to the wall on the opposite side of the aerosol source, the larger droplets are collected into the drain solution at the bottom of the atomizer. From the atomizer, the ACC aerosol mist passes through a diffusion-drying chamber at a flow rate of 1.1 l min−1 where the water vapour is rapidly absorbed (∼2 minutes residence). The drier, which is filled with a desiccant (silica gel), is 50 cm in length and 7.5 cm in diameter, consisting of an inner water vapour permeable tube (∼6 mm diameter) in which the aerosols pass through without direct contact with the desiccant. In addition, any particles, which hit the walls of the tubing, are lost from the gas flow hence the deposited ACC remain clean. EDX elemental analysis (Fig. S1, ESI†) on the deposited ACC and the absence of absorption bands characteristic of organic compound in the IR spectra confirmed the purity of the ACC.
The size distribution of the dry ACC is controlled by the initial concentration of the calcium hydroxide solution used. By using a condensation particle counter (CPC) the particle size distribution can be established and calibrated with respect to the calcium hydroxide solution. The particle size distribution Fig. 2 and the plot in (Fig. S2, ESI†) of the mean particle diameter as a function of concentration shows that using a 20 mM calcium hydroxide solution results in bigger ACC (mean size: ∼100 nm) whereas a 1 mM calcium hydroxide results in smaller ACC particles (mean size ∼60 nm). In addition, scanning electron microscopy (SEM) images in Fig. 3 (also Fig. S3, ESI†) shows that ACC generated from low calcium hydroxide concentration are well dispersed on the surface and do not agglomerate possibly due to their much smaller size. However, for higher concentrations, the ACC particles are much bigger in size thereby tend to quickly agglomerate. Control over ACC size distribution and agglomeration is therefore tuneable simply by adjusting the calcium hydroxide solution concentration and sample collection time.
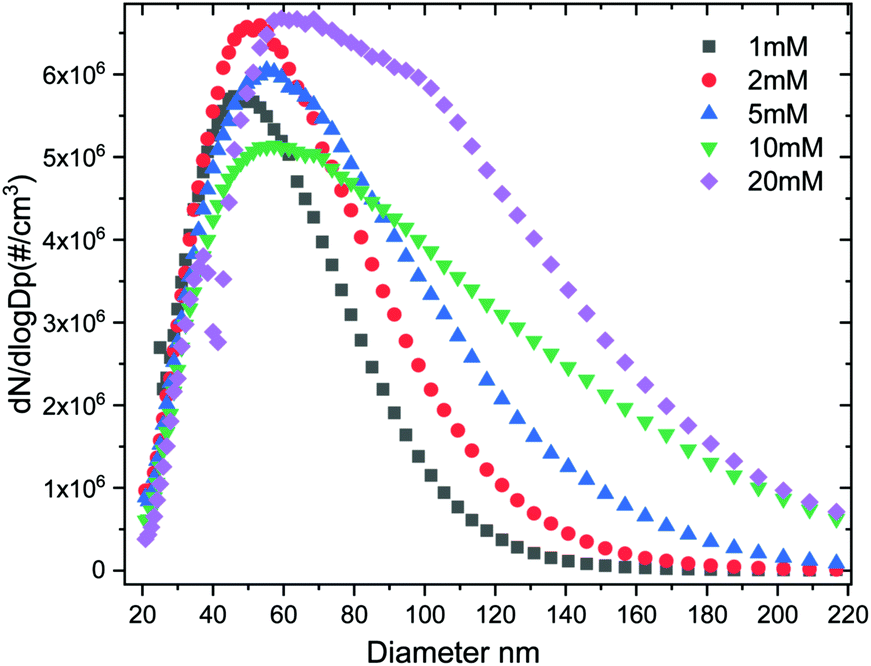 | ||
| Fig. 2 Number size distribution of aerosol ACC generated from different concentrations of calcium hydroxide. | ||
 | ||
| Fig. 3 SEM images of aerosol ACC prepared from (a) a 2 mM Ca(OH)2 solution and (b) a 20 mM Ca(OH)2 solution (scale bar 100 nm). | ||
The Ca K-edge XANES features of the aerosol-ACC in Fig. 4 are consistent with literature,14,31 displaying a pre-peak at ∼4039 eV similar to that in vaterite indicating the ACC structure is non centrosymmetric. The single broad white line at 4049 eV distinguishes the spectrum of the ACC from that of vaterite, which has a shoulder peak at ∼4044.5 eV, a white line peak at ∼4047.9 eV and a post edge feature centred at ∼4056 eV. The ACC XANES spectrum is also distinctively different from that of calcite, which displays a doubly split pre-edge feature at ∼4038.6 eV and ∼4039.8 eV, a shoulder peak at ∼4044 eV, a white line peak at ∼4047.3 eV and a further post edge feature centred at 4060 eV. The Ca K-edge XANES spectrum of calcium hydroxide on the other hand lacks a distinct pre-edge feature and is characterized by a shoulder peak at ∼4043.5 eV, a white line peak at ∼4050 eV and a post edge features which is in stark contrast to the ACC spectrum confirming the successful carbonation of the Ca(OH)2. The IR spectrum in Fig. 5 of the aerosol ACC also compares well with documented IR spectra of ACC.14,15,35,36 It is characterized by a doubly split asymmetric carbonate ion stretch at 1482 and 1420 cm−1 (ν3) indicative of lack of symmetry around the carbonate ion, a peak at 1073 cm−1 (ν1), an out-of-plane bending vibration at 864 cm−1 (ν2) and a broad in-plane bending vibration centred at 690 cm−1 (ν4).37 A broad O–H broad stretching vibration between 2700 and 3600 cm−1 and H–O–H bending vibration at 1650 cm−1 indicative of bound structural water, which is characteristic of ACC.
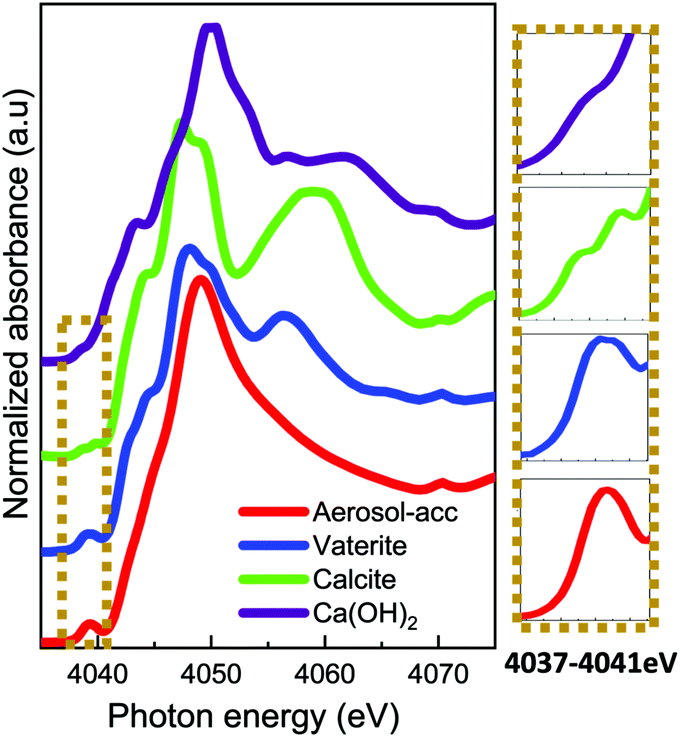 | ||
| Fig. 4 Ca K-edge XANES spectra of aerosol ACC, vaterite, calcite and calcium hydroxide. | ||
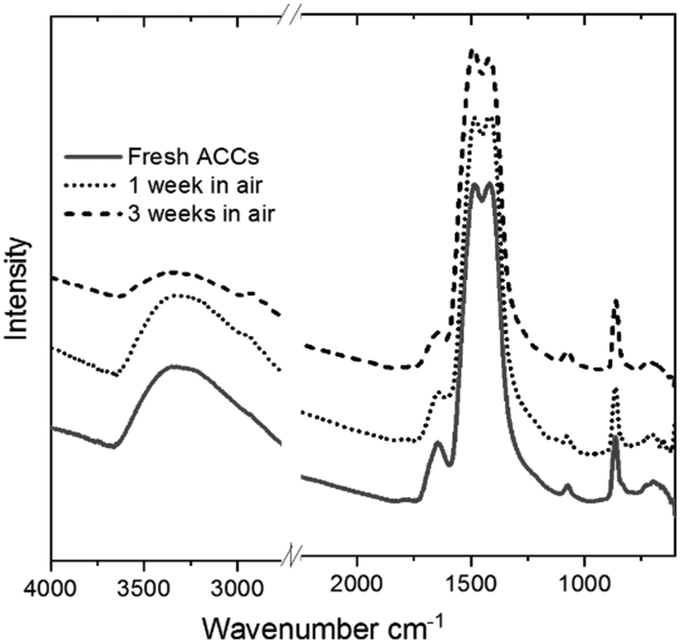 | ||
| Fig. 5 IR spectrum of freshly prepared dry aerosol ACC and exposed to air for 1 and 3 weeks. | ||
The significant absence of a sharp peak at 3640 cm−1 is a further confirmation of the complete carbonation of calcium hydroxide.38 When left exposed to air (∼33% relative humidity), the aerosol ACC show remarkable stability against crystallization with no observable difference noted in the IR spectra of the ACC even after 3 weeks in air. Upon exposure to very high relative humidity, a variety of ACC have been reported to rapidly crystallize to vaterite or calcite.35,39,40 However, at a relative humidity of up to ∼90%, the aerosol ACC display remarkable stability for extended periods. Fig. 6a shows time-resolved in situ Ca K-edge XANES measurements collected for 22 hours with continuous exposure of the aerosol ACC to 85% relative humidity. No evident changes in the spectra of the ACC are noted indicating their enhanced stability. However, despite the fact that there are no evident peaks characteristic of vaterite at 745 cm−1 or calcite at 713 cm−1 after 8 days of continuous ∼90% relative humidity exposure, some structural changes are observed in the IR spectra in Fig. 6b (also XANES in Fig. S4 ESI†). There is a broadening of the peak at 864 cm−1 with a small shift to higher wavenumbers and the broad O–H stretching vibration between 2700 and 3600 cm−1 increases in intensity whereas the H–O–H bending vibration at 1650 cm−1 decreases in intensity. This suggests that water induced structural changes occur in the aerosol ACC after several days of very high humidity exposure.
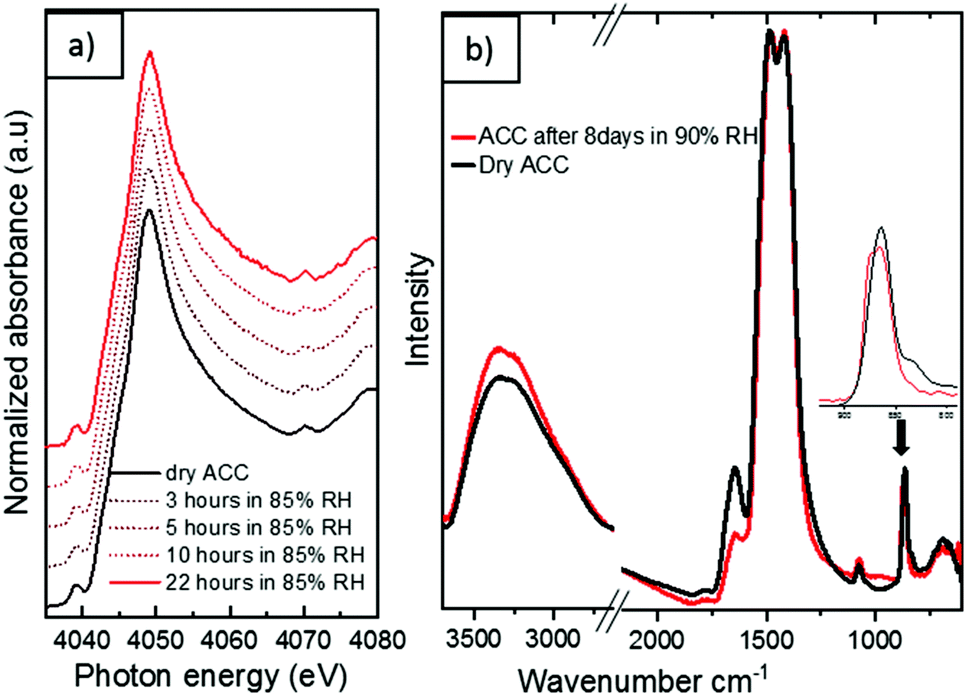 | ||
| Fig. 6 (a) In situ Ca K edge XANES of aerosol ACC during humidification with 85% RH for 22 hours (b) IR spectra of aerosol ACC before and after 8 days of 90% relative humidity (RH) exposure. | ||
We hypothesize that the enhanced stability of the aerosol ACC could be due to their purity and low water content arising from the rapid airborne drying. Most ACC synthesis procedures usually involve mixing two aqueous solutions, thereby traces of both salts from the counter ions,32 and water are inevitable. This hypothesis is consistent with observations from counter ion free freeze-dried ACC synthesized by Ihli et al.,27 which also exhibit enhanced stability for up to six weeks when left exposed to air. In addition, it has been proposed that the presence of mobile water in the ACC accelerates its crystallization,31,32,40,41 herein we rapidly dry individual airborne ACC before deposition on the sample holder thereby decreasing the probability of having trapped mobile water within the ACC aggregates upon deposition.
The aerosol-based synthesis of ACC presents a facile and reproducible approach of synthesizing impurity-free stable ACC.
These pure ACC can be used as references during studies on the structure and transformation of ACC prepared in the presence of counter ions (Na+/Cl−), impurities such as Mg2+ and PO42− ions or using other solvents such as ethanol and isopropanol. Having a consistent and reproducible reference will ultimately benefit the ongoing research on factors that control the crystallization pathway of ACC. In addition, other fields, such as bio-inspired material synthesis/drug delivery that rely on the purity and stability ACC may profit from this synthesis approach.
We thank the SNF (200021_157148) for financial support, the PHOENIX beamline at the Swiss Light source (Villigen, PSI) for beam time and Prof. Dr M. Ammann and Dr Peter Aaron Alpert for provision of a CPC and for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Kim, S. Choi, S. Kim, S. Lee, Y. Park and R. Chang, Calcium Carbonate Precipitation for CO2 Storage and Utilization: A Review of the Carbonate Crystallization and Polymorphism, Front. Energy Res., 2017, 5, 1–12 Search PubMed.
- Intergov. Panel Clim. Chang., ed. B. Metz, O. Davidson, H. de Coninck, M. Loos and L. Meyer, 2009, pp. 1–31 Search PubMed.
- W. Sekkal and A. Zaoui, Nanoscale analysis of the morphology and surface stability of calcium carbonate polymorphs, Sci. Rep., 2013, 3, 1587 CrossRef CAS PubMed.
- G. S. Dieckmann, G. Nehrke and C. Uhlig, Brief Communication: Ikaite (CaCO3·6H2O) discovered in Arctic sea ice, Cryosph., 2010, 227–230 CrossRef.
- J. De Yoreo, More than one pathway, Nat. Publ. Gr., 2013, 12, 284–285 CAS.
- R. Carlos, K. Kudłacz, Ö. Cizer and R. Encarnacion, Formation of amorphous calcium carbonate and its transformation into mesostructured calcite, CrystEngComm, 2015, 17, 58–72 RSC.
- M. Faatz, F. Gröhn and G. Wegner, Amorphous calcium carbonate: Synthesis and potential intermediate in biomineralization, Adv. Mater., 2004, 16, 996–1000 CrossRef CAS.
- C. Rodriguez-Navarro, A. Burgos Cara, K. Elert, C. V. Putnis and E. Ruiz-Agudo, Direct Nanoscale Imaging Reveals the Growth of Calcite Crystals via Amorphous Nanoparticles, Cryst. Growth Des., 2016, 16, 1850–1860 CrossRef CAS.
- J. D. Rodriguez-Blanco, S. Shaw and L. G. Benning, The kinetics and mechanisms of amorphous calcium carbonate (ACC) crystallization to calcite, via vaterite, Nanoscale, 2011, 3, 265–271 RSC.
- R. T. Devol, C. Sun, M. A. Marcus, S. N. Coppersmith, S. C. B. Myneni and P. U. P. A. Gilbert, Nanoscale Transforming Mineral Phases in Fresh Nacre, J. Am. Chem. Soc., 2015, 137, 13325–13333 CrossRef CAS PubMed.
- D. Pontoni, J. Bolze, N. Dingenouts, T. Narayanan, M. Ballauff and V. Uni, Crystallization of Calcium Carbonate Observed In situ by Combined Small- and Wide-angle X-ray Scattering, J. Phys. Chem., 2003, 5123–5125 CrossRef CAS.
- A. Navrotsky, Energetic clues to pathways to biomineralization: Precursors, clusters, and nanoparticles, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12096–12101 CrossRef CAS PubMed.
- E. M. Pouget, P. H. H. Bomans, J. A. C. M. Goos, P. M. Frederik, I. W. De and N. A. J. Sommerdijk, CaCO3 Formation Revealed by Cryo-TEM, Science, 2009, 323, 1455–1458 CrossRef CAS PubMed.
- B. Y. Politi, Y. Levi-kalisman, S. Raz, F. Wilt, L. Addadi, S. Weiner and I. Sagi, Structural Characterization of the Transient Amorphous Calcium Carbonate Precursor Phase in Sea Urchin Embryos, Adv. Funct. Mater., 2006, 16, 1289–1298 CrossRef.
- D. Gebauer, P. N. Gunawidjaja, J. Y. P. Ko, Z. Bacsik, B. Aziz, L. Liu, Y. Hu, L. Bergström, C. W. Tai, T. K. Sham, M. Edén and N. Hedin, Proto-calcite and proto-vaterite in amorphous calcium carbonates, Angew. Chem., Int. Ed., 2010, 49, 8889–8891 CrossRef CAS PubMed.
- M. Farhadi-khouzani, D. M. Chevrier, P. Zhang, N. Hedin and D. Gebauer, Amorphous Calcium Carbonate Water as the Key to Proto-Aragonite, Angew. Chem., Int. Ed., 2016, 8117–8120 CrossRef CAS PubMed.
- Y. Ma, B. Aichmayer, O. Paris, P. Fratzl, A. Meibom, R. A. Metzler, Y. Politi, L. Addadi, P. U. P. A. Gilbert and S. Weiner, The grinding tip of the sea urchin tooth exhibits exquisite control over calcite crystal orientation and Mg distribution, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6048–6053 CrossRef CAS PubMed.
- M. Eder, S. Amini and P. Fratzl, Biological composites—complex structures for functional diversity, Science, 2018, 362, 543–547 CrossRef CAS PubMed.
- D. M. Duffy, Coherent nanoparticles in calcite, Science, 2017, 358, 1254–1255 CrossRef CAS PubMed.
- J. T. Avaro, C. Ruiz-Agudo, E. Landwehr, K. Hauser and D. Gebauer, Impurity-free amorphous calcium carbonate, a preferential material for pharmaceutical and medical applications, Eur. J. Mineral., 2019, 31, 231–236 CrossRef.
- S. Matsumura, T. Sakamoto, T. Nishimura, B. Cantaert, T. Kato and D. Kuo, Use of Amorphous Calcium Carbonate for the Design of New Materials, ChemPlusChem, 2016, 82, 107–120 Search PubMed.
- U. G. K. Wegst, H. Bai, E. Saiz, A. P. Tomsia and R. O. Ritchie, Bioinspired structural materials, Nat. Mater., 2015, 14, 23–36 CrossRef CAS PubMed.
- J. W. Smith and R. J. Saykally, Soft X-ray Absorption Spectroscopy of Liquids and Solutions, Chem. Rev., 2017, 117, 13909–13934 CrossRef CAS PubMed.
- S. Weiner and L. Addadi, Crystallization Pathways in Biomineralization, Annu. Rev. Mater. Res., 2011, 41, 21–40 CrossRef CAS.
- S. Chen, H. Colfen, M. Antonietti and S. Yu, Ethanol assisted synthesis of pure and stable amorphous calcium carbonate nanoparticles, Chem. Commun., 2013, 49, 9564–9566 RSC.
- N. Koga, Y. Nakagoe and H. Tanaka, Crystallization of amorphous calcium carbonate, Thermochim. Acta, 1998, 318, 239–244 CrossRef CAS.
- J. Ihli, A. N. Kulak and F. C. Meldrum, Freeze-drying yields stable and pure amorphous calcium carbonate (ACC), Chem. Commun., 2013, 49, 3134–3136 RSC.
- Y. Politi, D. R. Bachelor, P. Zaslansky, B. F. Chmelka, J. C. Weaver, I. Sagi, S. Weiner and L. Addadi, Role of Magnesium Ion in the Stabilization of Biogenic Amorphous Calcium Carbonate: A Structure – Function Investigation, Chem. Mater., 2009, 22, 161–166 CrossRef.
- E. Loste, R. M. Wilson, R. Seshadri and F. C. Meldrum, The role of magnesium in stabilising amorphous calcium carbonate and controlling calcite morphologies, J. Cryst. Growth, 2003, 254, 206–218 CrossRef CAS.
- T. Roncal-Herrero, P. Bots, S. Shaw, J. D. Rodriguez-Blanco and L. G. Benning, The role of pH and Mg on the stability and crystallization of amorphous calcium carbonate, J. Alloys Compd., 2011, 536, S477–S479 Search PubMed.
- H. Du, M. Steinacher, C. Borca, T. Huthwelker, A. Murello, F. Stellacci and E. Amstad, Amorphous CaCO3: Influence of the Formation Time on Its Degree of Hydration and Stability, J. Am. Chem. Soc., 2018, 140, 14289–14299 CrossRef CAS PubMed.
- M. Albéric, L. Bertinetti, Z. Zou, P. Fratzl, W. Habraken and Y. Politi, The Crystallization of Amorphous Calcium Carbonate is Kinetically Governed by Ion Impurities and Water, Adv. Sci., 2018, 5, 1701000 CrossRef PubMed.
- Z. Zou, L. Bertinetti, Y. Politi, A. C. S. Jensen, S. Weiner, L. Addadi, P. Fratzl and W. J. E. M. Habraken, Opposite Particle Size Effect on Amorphous Calcium Carbonate Crystallization in Water and during Heating in Air, Chem. Mater., 2015, 27, 4237–4246 CrossRef CAS.
- B. Y. H. Liu and K. W. Lee, An aerosol generator of high stability, Am. Ind. Hyg. Assoc. J., 1975, 36, 861–865 CrossRef CAS PubMed.
- S. F. Chen, H. Cölfen, M. Antonietti and S. H. Yu, Ethanol assisted synthesis of pure and stable amorphous calcium carbonate nanoparticles, Chem. Commun., 2013, 49, 9564–9566 RSC.
- R. S. K. Lam, J. M. Charnock, A. Lennie, C. Fiona, R. S. K. Lam, J. M. Charnock and F. C. Meldrum, Synthesis-dependant structural variations in amorphous calcium carbonate, CrystEngComm, 2007, 9, 1226–1236 RSC.
- L. Addadi, S. Raz and S. Weiner, Taking advantage of disorder: Amorphous calcium carbonate and its roles in biomineralization, Adv. Mater., 2003, 15, 959–970 CrossRef CAS.
- M. Khachani, A. El Hamidi, M. Halim and S. Arsalane, Non-isothermal kinetic and thermodynamic studies of the dehydroxylation process of synthetic calcium hydroxide Ca(OH)2, J. Mater. Environ. Sci., 2014, 5, 615–624 CAS.
- X. Xu, J. T. Han, D. H. Kim and K. Cho, Two modes of transformation of amorphous calcium carbonate films in air, J. Phys. Chem. B, 2006, 110, 2764–2770 CrossRef CAS PubMed.
- F. Konrad, F. Gallien, D. E. Gerard and M. Dietzel, Transformation of Amorphous Calcium Carbonate in Air, Cryst. Growth Des., 2016, 16, 6310–6317 CrossRef CAS.
- H. Du and E. Amstad, Water: How does it influence the CaCO3 formation?, Angew. Chem., 2019 DOI:10.1002/ange.201903662.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional SEM images and particle size distribution. See DOI: 10.1039/c9cc03749g |
| This journal is © The Royal Society of Chemistry 2019 |