
Intramolecular palladium(II)/(IV) catalysed C(sp3)–H arylation of tertiary aldehydes using a transient imine directing group†‡
Sahra
St John-Campbell
 and
James A.
Bull
*
and
James A.
Bull
*
Department of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, Wood Lane, W12 0BZ, UK. E-mail: j.bull@imperial.ac.uk
First published on 8th July 2019
Abstract
Palladium catalysed β-C(sp3)–H activation of tertiary aldehydes using a transient imine directing group enables intramolecular arylation to form substituted indane-aldehydes. A simple amine bearing a methyl ether (2-methoxyethan-1-amine) is the optimal TDG to promote C–H activation and reaction with an unactivated proximal C–Br bond. Substituent effects are studied in the preparation of various derivatives. Preliminary mechanistic studies identify a reversible C–H activation, product inhibition and suggest that oxidative addition is the turnover limiting step.
Indanes display important and diverse biological activities, and occur in numerous marketed drugs (Scheme 1).1,2 Important developments in their synthesis include the use of transition metal catalysed intramolecular C–H functionalisation to cyclise relatively simple substrates bearing a halide, through reaction at a suitably located proximal C–H bond.3–6 To date this has been achieved using a Pd0/PdII redox cycle where regioselective C–H activation occurs following oxidative addition to a C–X bond by a Pd0 catalyst. Baudoin developed an enantioselective annulation using a chiral phosphine ligand and Pd(OAc)2 (Scheme 1a)3 and later developed a more general approach for the formation of 1-indanols and 1-indanamines (Scheme 1b).4 In the only example with an alkyl halide, Alexanian showed that oxidative addition to unactivated alkyl iodides could promote proximal C(sp2)–H activation and cyclisation to form 2,2- or 3,3-disubstituted indanes (Scheme 1c).5 Martin later merged oxidative addition, C(sp3)–H activation, and carbenoid insertion for Pd0/PdII catalysed indane formation (Scheme 1d).6
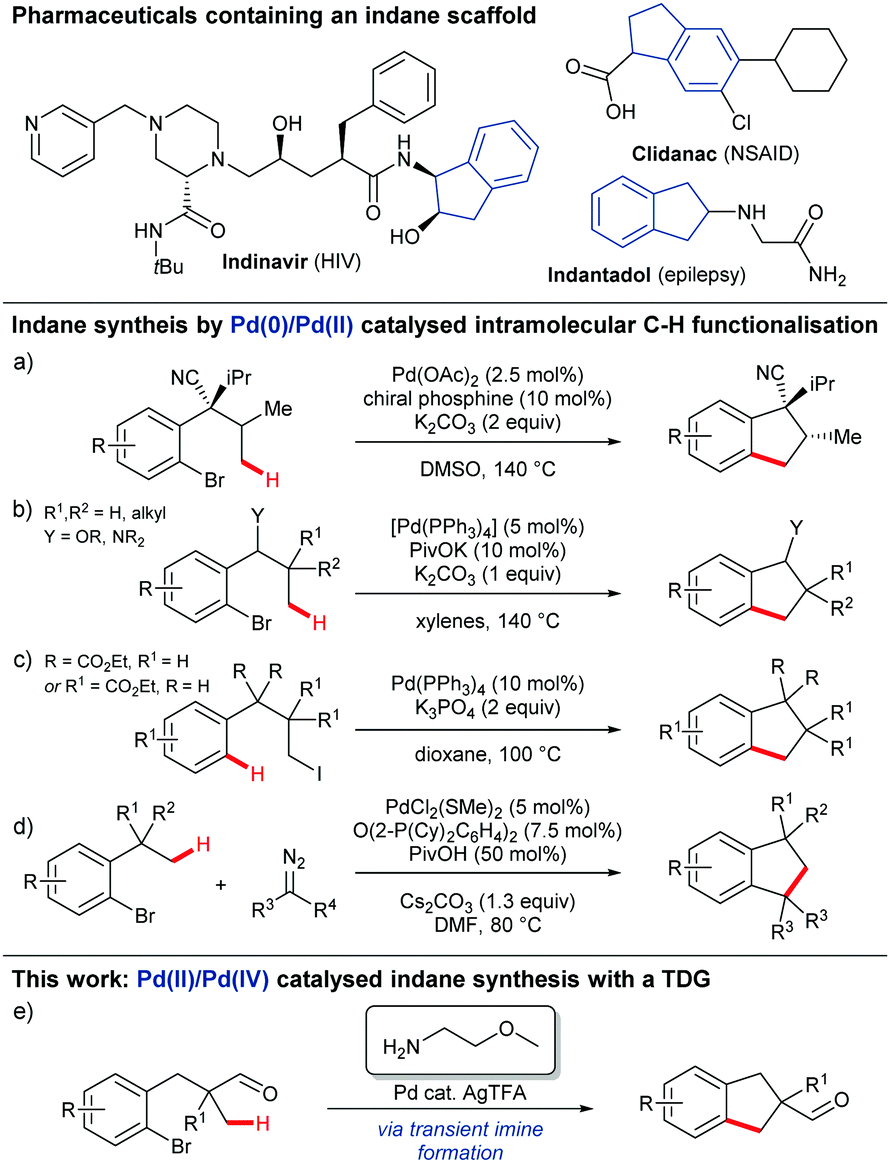 | ||
| Scheme 1 Indane synthesis using C–H functionalisation strategies. | ||
Here we disclose the first PdII/PdIV catalysed C–H arylation for indane synthesis where the C–H activation precedes the oxidative addition, using a transient directing group (Scheme 1e). The use of in situ generated and catalytic transient imine directing groups has recently emerged as an efficient strategy for directed C(sp3)–H functionalisation of aldehydes/ketones and amines to avoid the discrete steps for installation and removal of the directing groups.7–9 This is the first example of using a transient directing for intramolecular C(sp3)–H arylation and also the first example using unactivated aryl bromides.10
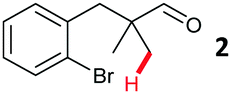
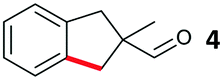
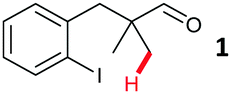
The model aryl halide containing substrates for intramolecular arylation (1–3) were readily synthesised in one step from commercial materials. The iodo, bromo and chloro substrates were subjected to conditions previously reported for the intermolecular arylation of tertiary aldehydes using N-tosylethylenediamine as a transient directing group (TDG1; Table 1).7c Iodide 1 cyclised to form indane 4 in 38% yield. Pleasingly, aryl bromide substrate 2 could also be cyclised under these conditions with better mass recovery than the iodide. The aryl chloride substrate 3 was unreactive.

Further investigation was undertaken with bromide 2, due to reduced degradation and to be more broadly tolerant of conditions required for the synthesis of a range of possible substrates.11 An improved yield of indane 4 was obtained with a longer reaction time, in the absence of the DMSO additive and using a higher ratio of HFIP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AcOH (Table 1, entry 4). Next, several amines were screened in substoichiometric amounts to form the transient imine directing group, with varied second coordinating groups and backbone length (Scheme 2). Simple α- or β-amino acids gave poor yields in the cyclisation (TDG2 and TDG3). Taurine (TDG4) formed the product in good yield. Aromatic TDG's, anthranilic acid (TDG5)7d and orthanilic acid (TDG6)7e were ineffective at promoting the reaction. The highest yield of 47% was obtained with the simple and inexpensive methoxy ether TDG7. The comparable phenyl ether (TDG8) was inferior to the methoxy group. Reducing the flexibility of the ether in a THF ring in TDG9 gave a similar yield to TDG7. Homologating the backbone gave only 11% yield (TDG10). A monodentate imine formed from 4-phenylbutylamine (TDG11) or benzylamine (TDG12) promoted the reaction in low yield. Importantly, when no amine was present the cyclised product was not formed, supporting the transient imine mechanism. These studies continue to demonstrate the importance of the second coordinating group, along with the potential of varied structure types to be successful.7,8 It is clear that the second binding site need not be an acidic group, and neutral coordination is suitable. Using 0.25 or 1 equiv. of TDG7 gave the same product yield.
:AcOH (Table 1, entry 4). Next, several amines were screened in substoichiometric amounts to form the transient imine directing group, with varied second coordinating groups and backbone length (Scheme 2). Simple α- or β-amino acids gave poor yields in the cyclisation (TDG2 and TDG3). Taurine (TDG4) formed the product in good yield. Aromatic TDG's, anthranilic acid (TDG5)7d and orthanilic acid (TDG6)7e were ineffective at promoting the reaction. The highest yield of 47% was obtained with the simple and inexpensive methoxy ether TDG7. The comparable phenyl ether (TDG8) was inferior to the methoxy group. Reducing the flexibility of the ether in a THF ring in TDG9 gave a similar yield to TDG7. Homologating the backbone gave only 11% yield (TDG10). A monodentate imine formed from 4-phenylbutylamine (TDG11) or benzylamine (TDG12) promoted the reaction in low yield. Importantly, when no amine was present the cyclised product was not formed, supporting the transient imine mechanism. These studies continue to demonstrate the importance of the second coordinating group, along with the potential of varied structure types to be successful.7,8 It is clear that the second binding site need not be an acidic group, and neutral coordination is suitable. Using 0.25 or 1 equiv. of TDG7 gave the same product yield.
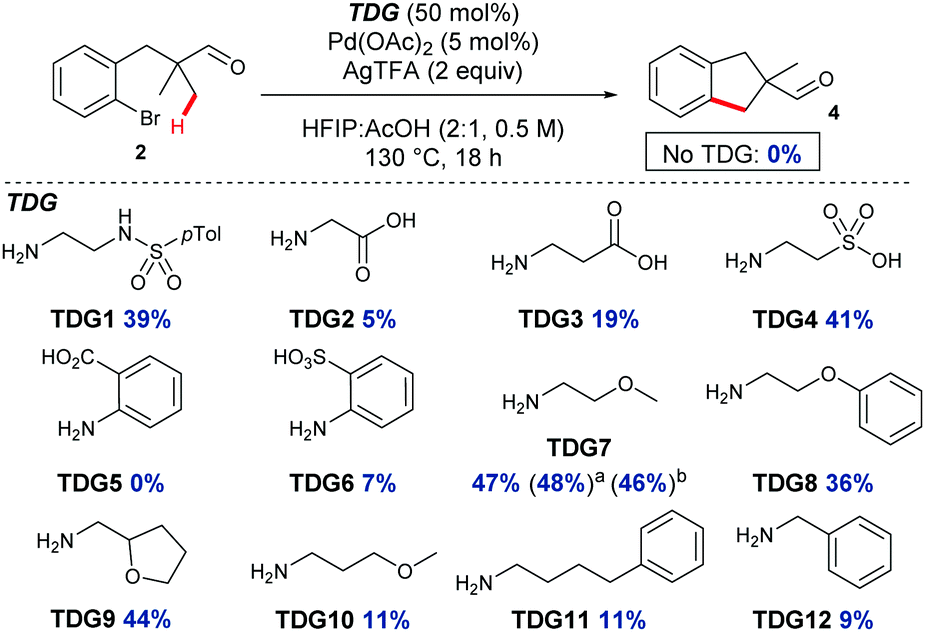 | ||
| Scheme 2 Screen of transient directing groups for C–H annulation, indicating the yield of indane 4. a25 mol% TDG7. b1 equiv. TDG7. | ||
A design-of-experiment (DOE) approach was employed to assess the continuous reaction parameters.11 The study did not ultimately lead to an improved yield of indane 4, but did provide additional insight. The reaction was successful (>25% yield) with high or low loadings of the TDG (0.25–0.75 equiv.), Pd(OAc)2 (2.5–10 mol%) and AgTFA (1–3 equiv.). A high concentration (1 M at 0.10 mmol 2) caused much decreased yields and poor mass recovery, particularly at low Pd loadings due to competing reactions. When lower concentrations were used (0.3 or 0.1 M in 2), a low 2.5 mol% loading of Pd(OAc)2 can achieve yields >45%. Notably, when using high loadings of TDG7 (0.75 equiv.), increased silver loadings gave better yields, but with less TDG7 (0.25 equiv.), a lower loading of AgTFA gave improved yields. This is likely to be as a consequence of α-oxidation of the amine by excess AgI. Although the yield remained modest due to unreacted 2, this constitutes the first example of intramolecular C(sp3)–H functionalisation using a transient directing group, as well as the first using unactivated aryl bromides.
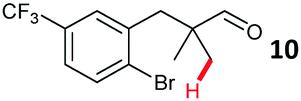
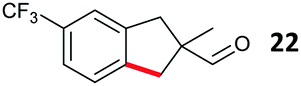
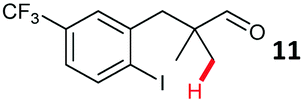
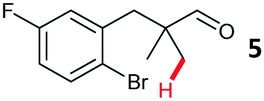
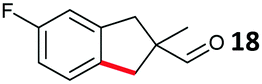
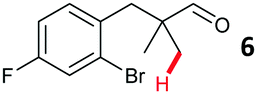
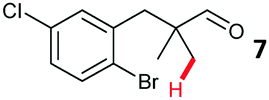
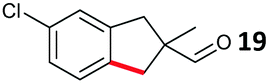
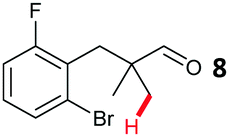
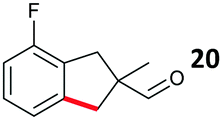
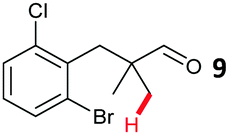
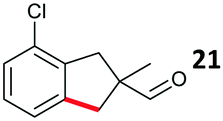
The study progressed to investigate the effects of different substituents on the aryl ring and alkyl group (Table 2). Indane 4 was isolated in a 42% yield from aryl bromide 2. On larger (2.0 and 4.0 mmol) scales, 4 was isolated in slightly reduced yields, with losses related to poor separation from the starting material. Iodo-substrate 1 was less effective under these conditions, with other unknown side products being formed. Interestingly, aldehyde 4 has previously been investigated for its odour properties.12 5-Fluoroindane 18 was accessed in the same isolated yield from either the 4-, or 5-fluoro precursors. 5-Chloroindane 19 was formed in good yield from the 5-substituted precursor 7, even at a larger 2.0 mmol scale. A chloride or fluoride in the 6-position was well tolerated for the cyclisation, however these substituents led to very little difference in polarity of between the starting material and cyclised product, and so were isolated as a mixture. 5-Trifluoromethyl indane 22 was isolated from both the bromide and iodide starting material, with the bromide again giving an improved yield.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Starting material | Product | Yield (%) | Entry | Starting material | Product | Yield (%) |
| a Yield of unreacted starting material isolated as a mixture with the product. Both yields calculated by the ratios of the aldehyde signals in the 1H NMR. | |||||||
| 1 |
|
|
42 | 8 |
|
|
42 |
| 34 (2 mmol) | |||||||
| 33 (4 mmol) | |||||||
| 2 |
|
20 | 9 |
|
28 | ||
| 3 |
|
|
41 | 10 |
|
|
5 |
| 4 |
|
42 | 11 |
|
26 | ||
| 5 |
|
|
46 47 (2 mmol) | 12 |
|
|
26 |
| 6 |
|
|
38 (16)a | 13 |
|
|
23 |
| 7 |
|
|
53 (10)a | 14 |
|
|
0 |

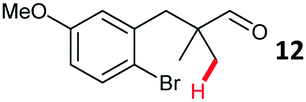
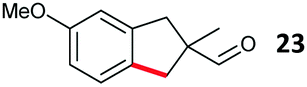
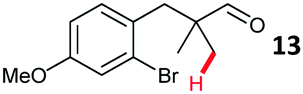
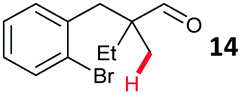
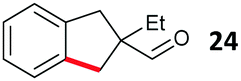
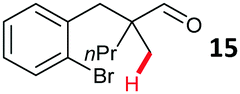
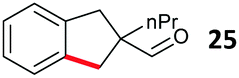
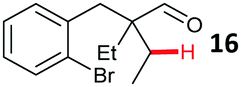
Aryl bromides with electron donating substituents were more challenging due to the already difficult oxidative addition of PdII to the C–Br bond. This was clearly noticeable with 5-methoxy substituent (12), where low conversions were observed, and only a 5% isolated yield of 23 was obtained. On the other hand, the same product could be accessed in higher yield when the electron donating group was moved to the meta-position with respect to the bromide (13), with a reduced influence on the oxidative addition. Larger alkyl substituents led to sequentially lower conversions, to afford indane aldehydes 24 and 25 in 26% and 23% yield respectively. Larger groups in this position suppressed cyclisation. Similarly, more electron rich or sterically hindered aromatics were not suitable. Substrates with only methylene C–H bonds accessible, such as 16, were not successful. Secondary aldehydes underwent preferential oxidation to the conjugated α,β-unsaturated aldehyde. Attempted cyclisation to other ring sizes (4-, 6- and 7-membered rings) gave trace product.11
Indane-aldehydes were derivatised to demonstrate their synthetic utility (Scheme 3). Indane 4 was readily reduced to alcohol 27 in excellent yield, and reductive amination with morpholine furnished amine 28. The addition of methyl magnesium chloride to methoxy-indane 23 gave secondary alcohol 29. Finally, we functionalised the remaining methyl group of chloride containing aldehyde 19 by an intermolecular transient C–H functionalisation using our previously reported protocol with 4-iodoanisole, to give arylated indane 30.7c
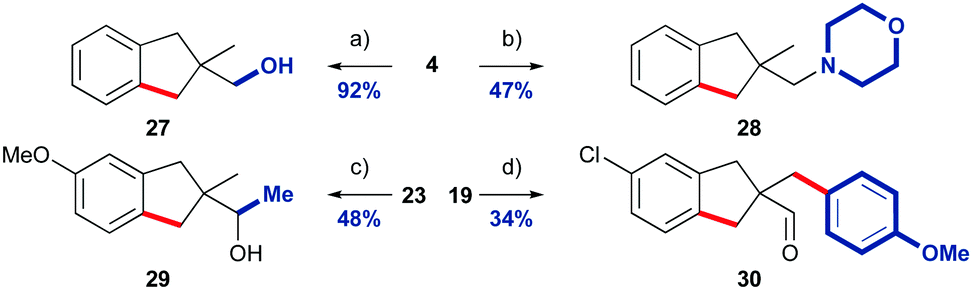 | ||
| Scheme 3 Derivatisation of indane products. (a) NaBH4, MeOH, 2 h 0–25 °C, (b) morpholine, NaBH(OAc)3, CH2Cl2, 25 °C, 24 h. (c) MeMgCl, Et2O, 0–25 °C, 1.5 h. (d) TDG1 (50 mol%), 4-iodoanisole, Pd(OPiv)2 (5 mol%), AgTFA, DMSO (1 equiv.), HFIP:AcOH (1:1), 130 °C, 3 h. Product isolated as a 5.7:1 mixture with a 4-acetoxyanisole by-product. | ||
Studies were undertaken to provide insight to the mechanism of the intramolecular C–H arylation (Scheme 4). Performing the reaction in deuterated solvents gave β-deuteration of the methyl groups of both aldehyde 2 and indane-aldehyde 4 (Scheme 4a), suggesting reversible C–H activation, deuteration at other positions was not observed. Indane 4 was deuterated under the reaction conditions in absence of starting material (Scheme 4b), suggesting unproductive product cyclopalladation may occur in the reaction. On prolonged heating of the product, side reactions led to poor mass recovery. Competition experiments with an external aryl iodide (4-iodoanisole) using bromide substrate 2 revealed a mixture of products with preferential intermolecular arylation (Scheme 4c). Indane 4 was observed in only 7% conversion from the starting material, and the arylated product of the indane (31) was formed in 10%,13 whereas products of intermolecular arylation (mono + di) were obtained in 44% conversion indicating slow oxidative addition of the internal aryl bromide. In contrast, the intramolecular cyclisation was favoured with iodo-substrate 1, with 62% conversion to the intramolecular arylation products (Scheme 4d). The reaction profile obtained by running individual reactions at different time points indicated rapid, apparent zero-order reaction kinetics under the standard conditions with the majority of the product formed in only 2 h, after this the conversion plateaued suddenly.11,14,15 A ‘same excess’ kinetics study revealed that inhibition from the product was likely in the reaction.11,16 The higher observed deuterium incorporation in the product compared to the starting material could suggest a potential mechanism of product inhibition, through preferential unproductive reversible β-cyclopalladation of the imine derived from indane 4. Mechanistically we propose that aldehyde 2 and TDG7 first condense to form imine I which coordinates to the PdII catalyst (Scheme 4e). Concerted metallation deprotonation occurs (II) to activate the C–H bond and form palladacycle III. The aryl bromide is suitably located to coordinate to the Pd centre and undergo oxidative addition to give PdIV intermediate IV. Due to the pronounced effect of electron-rich aromatic groups on the yield, and observed zero-order reaction, the oxidative addition is likely to be the turnover limiting step. Reductive elimination furnishes indane imine V. Imine V can be cyclometallated at the methyl group, shown by competition and deuteration experiments, removing Pd from the productive cycle as VI.
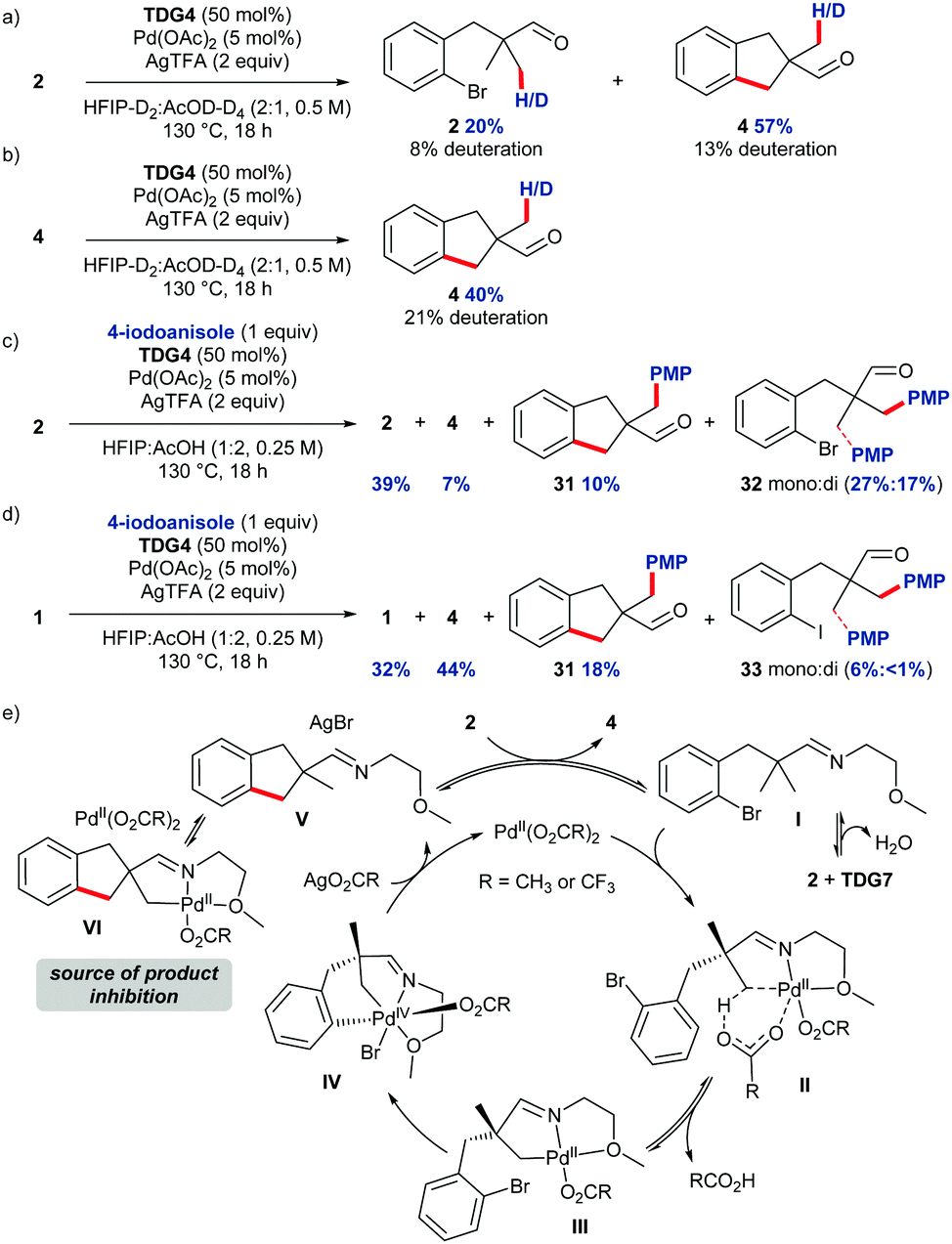 | ||
| Scheme 4 Mechanistic experiments. (a) Deuteration experiment showed small levels of deuteration in RSM 2 and indane 4, yields determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. (b) Deuteration of indane 4, yields determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. Competition experiments with (c) external aryl iodide and internal aryl bromide, and (d) external aryl iodide and internal aryl iodide, values given are conversions. (e) Proposed catalytic cycle. | ||
In conclusion, indanes can be formed by intramolecular C(sp3)–H arylation using a transient directing group. 2-Methoxyethan-1-amine can form a transient imine directing group to promote proximal β-C(sp3)–H activation of the cyclisation precursors and facilitate oxidative addition and subsequent cyclisation through a PdII/PdIV redox cycle. The product yields for this reaction remained relatively low, related to the difficult PdII oxidative addition and potential product inhibition. However, this presents the first intramolecular C(sp3)–H functionalisation with a TDG and the first use of unactivated aryl bromides using a TDG, and may stimulate further study. Various substituents on the aromatic ring and secondary alkyl groups were tolerated to prepare indane aldehydes, and the aldehyde substituent was also further derivatised. Preliminary mechanistic studies in the form of deuteration, competition and same excess experiments suggested reversible C–H activation, a turnover-limiting oxidative addition and product inhibition through unproductive cyclometallation of the product imine.
We gratefully acknowledge The Royal Society (University Research Fellowship UF140161, to JAB), and EPSRC (Doctoral Prize Fellowship to SSJC).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Vilums, J. Heuberger, L. H. Heitman and A. P. IJzerman, Med. Res. Rev., 2015, 35, 1097–1126 CrossRef CAS; (b) B. D. Dorsey, R. B. Levin, S. L. McDaniel, J. P. Vacca, J. P. Guare, P. L. Darke, J. A. Zugay, E. A. Emini and W. A. Schleif, J. Med. Chem., 1994, 37, 3443–3451 CrossRef CAS; (c) G. R. Allen, R. Littell, F. J. McEvoy and A. E. Sloboda, J. Med. Chem., 1972, 15, 934–937 CrossRef CAS; (d) G. Villetti, G. Bregola, F. Bassani, M. Bergamaschi, I. Rondelli, C. Pietra and M. Simonato, Neuropharmacology, 2001, 40, 866–878 CrossRef CAS.
- B. Gabriele, R. Mancuso and L. Veltri, Chem. – Eur. J., 2016, 22, 5056–5094 CrossRef CAS.
- N. Martin, C. Pierre, M. Davi, R. Jazzar and O. Baudoin, Chem. – Eur. J., 2012, 18, 4480–4484 CrossRef CAS.
- S. Janody, R. Jazzar, A. Comte, P. M. Holstein, J.-P. Vors, M. J. Ford and O. Baudoin, Chem. – Eur. J., 2014, 20, 11084–11090 CrossRef CAS.
- A. R. O. Venning, P. T. Bohan and E. J. Alexanian, J. Am. Chem. Soc., 2015, 137, 3731–3734 CrossRef CAS.
- Á. Gutiérrez-Bonet, F. Juliá-Hernández, B. de Luis and R. Martin, J. Am. Chem. Soc., 2016, 138, 6384–6387 CrossRef PubMed.
- For aldehydes, see: (a) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252–256 CrossRef CAS; (b) K. Yang, Q. Li, Y. Liu, G. Li and H. Ge, J. Am. Chem. Soc., 2016, 138, 12775–12778 CrossRef CAS; (c) S. St John-Campbell, A. J. P. White and J. A. Bull, Chem. Sci., 2017, 8, 4840–4847 RSC; (d) X.-H. Liu, H. Park, J.-H. Hu, Y. Hu, Q.-L. Zhang, B.-L. Wang, B. Sun, K.-S. Yeung, F.-L. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 888–896 CrossRef CAS; (e) X.-Y. Chen and E. J. Sorensen, J. Am. Chem. Soc., 2018, 140, 2789–2792 CrossRef CAS PubMed . For amines, see: ; (f) Y. Liu and H. Ge, Nat. Chem., 2016, 9, 26–32 CrossRef; (g) Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554–14557 CrossRef CAS; (h) S. St John-Campbell, A. K. Ou and J. A. Bull, Chem. – Eur. J., 2018, 24, 17838–17843 CrossRef CAS.
- For reviews on transient C–H functionalisation, see: (a) H. Sun, N. Guimond and Y. Huang, Org. Biomol. Chem., 2016, 14, 8389–8397 RSC; (b) Q. Zhao, T. Poisson, X. Pannecoucke and T. Besset, Synthesis, 2017, 4808–4826 CAS; (c) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS; (d) S. St John-Campbell and J. A. Bull, Org. Biomol. Chem., 2018, 16, 4582–4595 RSC.
- (a) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902–4911 RSC; (b) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS; (c) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS.
- Yu has shown that 2-pyridyl bromides can be suitable coupling partners with a TDG, see: Y.-Q. Chen, Z. Wang, Y. Wu, S. R. Wisniewski, J. X. Qiao, W. R. Ewing, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 17884–17894 CrossRef CAS.
- See ESI‡ for further details.
- B. Winter and S. Gallo-Flückiger, Helv. Chim. Acta, 2005, 88, 3118–3127 CrossRef CAS.
- Intermolecular arylation must proceed after cyclisation, as shown by the unsucessful cyclisation of substrate 16.
- Addition of further Pd(OAc)2, TDG7 or AgTFA after 3 h did not lead to further conversion to product 4.
- Comparison of the reaction profiles at 2.5 mol% and 5 mol% Pd(OAc)2 suggested the reaction was first order in Pd.
- (a) D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302–4320 CrossRef CAS; (b) R. D. Baxter, D. Sale, K. M. Engle, J.-Q. Yu and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 4600–4606 CrossRef CAS.
Footnotes |
| † All raw and processed data for this manuscript can be found at the Imperial College London Research Data Repository (DOI: http://10.14469/hpc/5614). |
| ‡ Electronic supplementary information (ESI) available: Additional optimisation and control reactions, DOE results, kinetic, competition and deuteration experiments, experimental procedures and characterisation data. See DOI: 10.1039/c9cc03644j |
| This journal is © The Royal Society of Chemistry 2019 |