
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEvolving polymersomes autonomously generated in and regulated by a semibatch pH oscillator†
Jinshan
Guo‡
 a,
Eszter
Poros-Tarcali‡
a and
Juan
Perez-Mercader
*ab
a,
Eszter
Poros-Tarcali‡
a and
Juan
Perez-Mercader
*ab
aDepartment of Earth and Planetary Science and Origin of Life Initiative, Harvard University, 20 Oxford Street, Cambridge, Massachusetts 02138, USA. E-mail: jperezmercader@fas.harvard.edu
bSanta Fe Institute, Santa Fe, New Mexico 87501, USA
First published on 18th July 2019
Abstract
A semibatch bromate–sulfite (B–S) pH oscillator is applied to drive the autonomous generation of polymersomes through polymerization induced self-assembly (pH-O-PISA) as a provider of radicals for polymerization of a hydrophobic monomer extending from a 2-sulfur hydrophilic macromolecular chain transfer agent (mCTA) to form an amphiphilic copolymer, and to regulate the morphological evolution of the self-assembled polymersomes.
Self-assembly is a spontaneous process governed by thermodynamically favourable interactions, during which a system from its building blocks constructs a highly organized structure by noncovalent bonds without any external intervention.1 In living systems, the presence of an interface, self-assembled from amphiphiles, separating the aqueous contents of individual cells from their external environment is essential. This has inspired the self-assembly of lipid-based amphiphiles to liposomes as the most studied artificial cell models.2,3 Vesicles self-assembled by amphiphilic copolymers are called polymersomes.4 Compared to lipid-based amphiphiles, amphiphilic copolymers possess higher molecular weights and allow a broader choice of chemistry, which not only endows polymersomes with enhanced stability, toughness, and tuneable physical and chemical properties, but also with various introduced functionalities for the development of stimuli (thermal, redox, light and pH) responsive and artificially engineered polymersomes.5–11 In addition, the technique of polymerization induced self-assembly (PISA)12–16 provides a facile one-pot approach for the de novo synthesis of polymersomes from simpler components without using complicated, multi-step procedures. Here the ongoing polymerization of the hydrophobic block onto the hydrophilic chain, and as the packing parameter of the amphiphiles changes, induces a dynamic morphology evolution.17 This “living” feature makes PISA-generated polymersomes especially interesting protocell models in top-down synthetic biology.18
Beside numerous other features, the cell membrane is known to show dynamic morphology changes triggered by the environment. To mimic this behavior, oscillatory shape deformations in colloidosomes,19 liposomes20 and polymersomes21–23 have been implemented using oscillatory reactions. Among the oscillatory chemical reactions, pH oscillators are one of the most versatile. They display large amplitude pH oscillations in unbuffered media.24 A feature that makes them great candidates for practical applications, which until now are all based on pH-sensitive processes coupled to pH oscillators.25–30 It is common in all pH oscillators, that the concentration of H+, as the autocatalytic species, plays a critical role in the kinetics of the systems. Most of them are based on redox chemistry and many of these reactions are known to proceed with the involvement of free radical steps.31 These lead us to the idea of coupling pH oscillators with PISA, expecting that pH oscillators will not only drive the RAFT polymerization via the generated radicals but also control the morphological evolution of the self-assembled structures generated with PISA.
In this communication, a semibatch bromate–sulfite (B–S) pH oscillator32,33 is coupled with PISA. All pH oscillators work under flow conditions, because one or more substrates are consumed in one period and need to be supplied to start a new period. Using a semibatch reactor, which has only an inlet and no outlet, instead of a CSTR (continuously fed stirred tank reactor), allows polymerization to proceed long enough so as to achieve high conversion and facilitate the investigation of morphological evolution by trapping all the self-assembled structures into a closed vessel. In our semibatch reactor the solution of sodium sulfite and sulfuric acid was continuously inflowed to the mixture of sodium bromate, butyl-acrylate (BA) and poly(ethylene glycol) 2-sulfur chain transfer agent (PEG-CTA) molecules (Scheme 1). The chemical energy and radicals generated by pH oscillatory chemistry were used to drive the polymerization of hydrophobic BA monomers extending from one end of the hydrophilic PEG-CTA molecules and control the concurrent self-assembly (pH-O-PISA, pH oscillator-driven and controlled polymerization induced self-assembly) of the amphiphilic block copolymer, poly(ethylene glycol)-block-poly(butyl acrylate) (PEG-b-PBA) (Scheme 1).
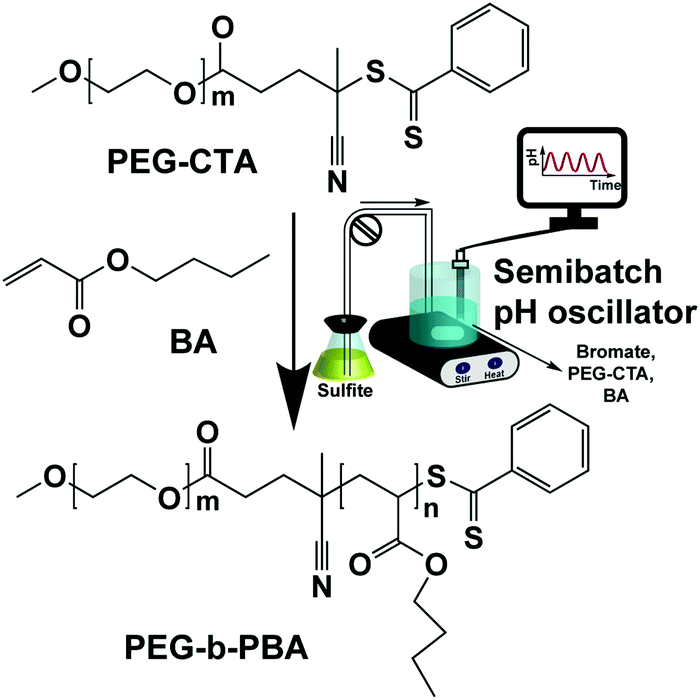 | ||
| Scheme 1 Synthesis of amphiphilic PEG-b-PBA block copolymers using radicals generated by a semibatch bromate–sulfite (B–S) pH oscillator. | ||
For PEG-b-PBA with different target degrees of polymerization (DPtargets, equal to the feeding ratios of monomer/CTA), the pH oscillation curves in pH-O-PISA are shown in Fig. 1a (DPtarget = 100) and Fig. S1 (ESI†) (DPtarget = 75 and 150). For DPtarget = 100 (Fig. 1a), the ΔpH and pHmin values both stay in a narrow range between 3.4 and 3.8, while the periods of the pH oscillations increased from ∼20 to ∼45 minutes as the oscillations proceed (except for the first period, which is always different) (Fig. S2, ESI†). A similar increase in period time is also observed in pH-O-PISA of PEG-b-PBA with DPtargets of 75 and 150 (Fig. S1, ESI†). The consumption of bromate and monomer along with the dilution might contribute to this. As can be seen from Fig. S3 (ESI†), after the pH oscillations of the pH-O-PISA for PEG-b-PBA (DPtarget = 75) stopped, one more pH oscillation period was realized when additional monomer was added into the system, indicating that the polymerization is not only a consequence but also a driving force for the pH oscillations in pH-O-PISA for this particular composition. The B-S pH oscillatory reaction is very sensitive to H+ input. The inclusion of PEG-CTA and monomer made the original B–S pH oscillator (0.06 M H+ in the inflowed solution, Fig. S4a, ESI†) slip out of the oscillatory regime (Fig. S4b, ESI†). In order to restore the oscillatory behaviour in the PISA coupled system, the concentration of the inflowed H+ needs to be increased (0.185 M, using BA as monomer, Fig. 1a). Similarly, high H+ concentration (0.185 M) would bring the pure B–S system out of its oscillatory regime too (Fig. S5, ESI†), and at this composition the polymerization is necessary in order to bring the pH back from the acidic steady state (Fig. 1a and Fig. S1, S3, ESI†). Our pH-O-PISA system can be universally adapted to different monomers, such as 2-methoxyethyl methacrylate (MEMA) (Fig. S6, ESI†), by the adjustment of the H+ concentration in the inflowed Na2SO3–H2SO4 solution and flow rate.
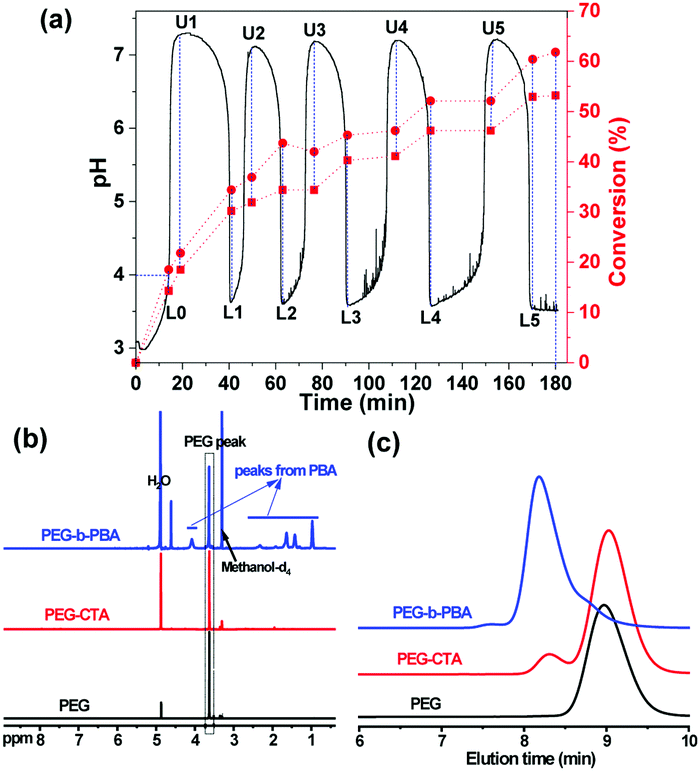 | ||
| Fig. 1 Polymerization driven and controlled by the pH oscillator: (a) pH oscillation curve in a semibatch B–S pH oscillator with the existence of PEG-CTA and butyl acrylate (BA) monomer at 40 °C; monomer conversions (calculated by the area ratios of A(–CH2OCO–)/A(PEG) (red square dashed line) and A(–CH2–CH2OCO–)/A(PEG) (red circle dish line)). 1H-NMR spectra (b) and GPC curves (c) of PEG (DP ∼ 43), PEG-CTA, and purified PEG-b-PBA collected after 180 min. | ||
The polymerization process of BA was studied through the 1H-NMR tests of the freeze-dried reaction mixtures sampled at critical and preset time points (labelled in Fig. 1a, where LX denotes low point X and UX upper point X). The molecular weight of the purified final PEG-b-PBA block copolymer was determined both by 1H-NMR and GPC (Fig. 1 and Table S1, ESI†). The monomer conversion (%) was calculated in two ways: (1) by the area ratio of A(–CH2OCO–)/A(PEG) (–CH2OCO– refers to the proton near the ester bond with a chemical shift ∼4.2 ppm, red square dashed line in Fig. 1a); (2) based on the area ratio of A(–CH2–CH2OCO–)/A(PEG) (the chemical shift of –CH2–CH2OCO– is at ∼1.65 ppm, red circle dashed line in Fig. 1a). The monomer peak cannot be observed in the NMR spectra because the residual monomers are removed by freeze-drying before the tests (Fig. S7, ESI†). The monomer conversion of BA gradually increases in time as the pH oscillations proceed (Fig. 1a and Fig. S7, ESI†), as can be clearly seen from the increase in the areas of the PBA peaks (labelled in Fig. 1b) referring to the constant area of the PEG peak in the 1H-NMR spectra of reaction mixtures sampled out at different times (Fig. S7, ESI†). Both monomer conversion (%) vs. time curves (Fig. 1a) indicate a fast polymerization when the pH decreases and a slower or negligible polymerization when the pH increases (especially so after the first two periods), indicating that the polymerization is periodic in pH-O-PISA.
The two-way oxidation of the sulfite by bromate serves as a radical source for the initiation of the polymerization. The autocatalytic complete oxidation of SO32− to SO42− generates H+ (reactions (3) and (4) in Table S2 (ESI†), positive feedback), and the partial oxidation of SO32− (1–2%) to S2O62− consumes H+ (reaction (5) in Table S2, negative feedback). The protonation of the constantly inflowed SO32− (reactions (1) and (2) in Table S2, ESI†) also contributes to the negative feedback, but in these reactions no radicals are formed. An appropriate time lag between the two loops results in periodic pH changes in the system, during which a periodic radical supply arises due to the difference in the overall reaction rates and the conversions (Table S2, ESI†) in the pH dropping and rising segments of each period.32
The final monomer conversion at 180 min calculated by the area ratio of A(–CH2OCO–)/A(PEG) (red square dashed line in Fig. 1a) was 53.2%; while based on the area ratio of A(–CH2–CH2OCO–)/A(PEG) (red circle dashed line in Fig. 1a), it was 61.8%. The DP of the purified final PEG-b-PBA product was 58.6 determined by 1H-NMR (Fig. 1b and Table S1, ESI†), and 50.7 determined by GPC (Fig. 1c and Table S1, ESI†), the latter being closer to the conversion calculated by the area ratio of A(–CH2OCO–)/A(PEG) of the final reaction mixture (53.2, Fig. 1a and Fig. S7, Table S1, ESI†). Monomer evaporation during pH-O-PISA (operated at 40 °C) might be one of the reasons why 100% monomer conversion was not achieved. The polydispersity index (PDI) of the final PEG-b-PBA was calculated to be 2.16 by GPC (Table S1, ESI†).
We thoroughly investigated the self-assembly process in pH-O-PISA of PEG-b-PBA with a DPtarget = 100. The size change of self-assembled structures along with pH oscillations was monitored by DLS (Fig. 2). The morphology of self-assembled structures at different times was observed by TEM/SEM, and/or cryo-SEM (Fig. 3 and 4). Fig. 2 shows how the size of the self-assembled structures increased in time from nanometer to micrometer scales with the progress of pH-O-PISA. Micelles with diameters <100 nm were observed at 30 min (Fig. 3a (TEM) and Fig. S8 (SEM), ESI†), then the micelles transformed into nano-sized vesicles with an average diameter of 138 nm at 60 min (Fig. 3b and Fig. S9, ESI†). The size of the vesicles increased to ∼1 μm at 120 min (Fig. 2 and 3c). Giant vesicles with sizes of one to tens of μm were formed after 150 min (Fig. 2, 3d, e1, e2 and Fig. S10, S11, ESI†). An unexpected decrease in size of the 180 min sample compared to the 150 min sample was observed in the DLS test (Fig. 2). This could be ascribed to the aggregation and precipitation of giant vesicles, as suggested by the appearance of a second but smaller peak at 5–50 μm in the DLS curve of the 180 min sample (Fig. 2). An energy dispersive spectroscopy (EDS) study of the unwashed 180 min sample proved the existence of salts derived from bromate and sulfite inside and outside the vesicles (Fig. S12, ESI†).
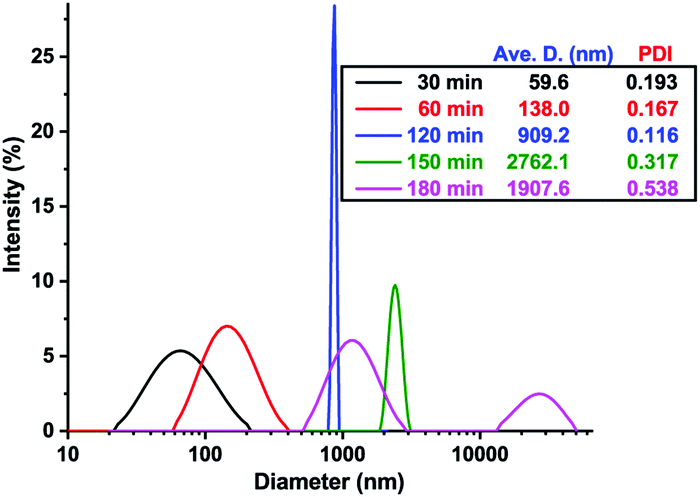 | ||
| Fig. 2 DLS result of pH-O-PISA of PEG-b-PBA (DPtarget = 100) samples sampled out at different time points. | ||
 | ||
| Fig. 3 T/SEM image of pH-O-PISA of PEG-b-PBA (DPtarget = 100) samples sampled out at different time points (a: 30 min, b: 60 min, c: 120 min, d: 150 min, e1 (unwashed), e2 (washed): 180 min). | ||
 | ||
| Fig. 4 Cryo-SEM images of the final (180 min) giant vesicles self-assembled by PEG-b-PBA obtained by pH-O-PISA (DPtarget = 100). | ||
To further confirm the formation of giant vesicles, a cryo-SEM test was conducted which can freeze and preserve the original morphology of vesicles dispersed in aqueous media. Freeze-fractured hollow structures with diameters >1 μm can be clearly seen from the cryo-SEM images shown in Fig. 4, especially Fig. 4c and d, thus proving the successful formation of giant vesicles in pH-O-PISA of PEG-b-PBA.
Evolution of the vesicles generated by pH-O-PISA was observed as vesicle budding and division of both small vesicles in the 60 min sample (Fig. 3a and Fig. S9, ESI†) and giant vesicles in the 180 min sample (Fig. 3e1, e2 and Fig. S10, S11, ESI†). Freeze-fractured vesicle division can also be seen from the cryo-SEM image of the 180 min sample (Fig. 4a). The presence of the vesicle membrane and the nature of pH oscillatory reactions coupled with the continuous inflow of sulfite can induce: (1) a chemical gradient across the vesicle membrane, (2) a chemical reaction difference in the inner and outer medium of the vesicle (the reactions inside the vesicles tend to decay from the pH oscillations of the outside solution because they are out of reach for the sulfite supply) and (3) a gradual increase of overall salt concentration. These factors make the self-assembled polymersomes always stay in a non-equilibrium environment, and evolve in morphology which results in the budding and division of the vesicles.34
Beside the above-mentioned effects, the increasing salt concentration together with the advance of the pH-O-PISA reaction might also play an additional role in the formation of giant vesicles. For fatty acid-based vesicles, decreasing critical vesicle concentration (CVC) with the increase of salt concentration has been reported.35 When salt was added to water, entropic forces become more favorable to exclude hydrophobic structures, resulting in lower solubility of the hydrophobic block compared to that in pure water. A morphology conversion from micelles to vesicles was also observed when salts were added to a medium containing amphiphilic copolymers.36 The gradual increase of salt concentration might contribute to increasing the “effective packing parameter” and promote the formation of giant vesicles.
In summary, we have demonstrated that the periodic generation of radicals by the semibatch B–S pH oscillator can initiate the polymerization of the hydrophobic monomer butyl acrylate (BA), from a 2-sulfur hydrophilic PEG macro-RAFT agent and the polymerization induces the cooperative self-assembly of the amphiphiles into collective objects. The transformation of these autonomously generated structures from nano-sized micelles to submicron then giant vesicles (up to 10 μm) is also controlled by our pH oscillator system. Furthermore, besides driving the polymerization, the pH oscillator provides additional controlling factors for the process of self-assembly. The constant inflow of sulfite in the semibatch reactor and the oscillations put the generated polymersomes in an out-of-equilibrium environment by generating a chemical gradient and chemical reaction difference between the inside and outside of the vesicles, contributing to the evolution of the self-assembled polymersomes into episodes of budding and division. Furthermore, the gradual increase in the salt concentration improves the “effective packing parameter” and promotes the formation of giant vesicles. The better understanding of these processes promises more chances for controlled vesicle evolution, including the possible self-replication, not just division. Finally, pH-O-PISA can be universally adaptable to different vinyl monomers, and the inflow of other reactants, such as macro-CTA and/or monomers, the use of a continuous stirred-tank reactor (CSTR), or the introduction of pH-responsive monomers are also possible to study in the future together with the combination of a pH oscillator with polymerization induced electrostatic self-assembly (PIESA).37
We thank Dr Gong Cheng and Dr Liman Hou for help with the TEM study, and Adam Graham from the Center for Nanoscale Systems (CNS) at Harvard University for the Cryo-SEM study. The DLS, TEM and SEM studies were conducted in the facilities of the CNS at Harvard University. This work was supported by Repsol S. A. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflicts of interest
There are no conflicts to declare.Notes and references
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS.
- A. Pohorille and D. Deamer, Trends Biotechnol., 2002, 20, 3 CrossRef.
- Y. Ding, F. Wu and C. Tan, Life, 2014, 4, 4 CrossRef.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 5583 CrossRef.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Land-fester, Chem. Soc. Rev., 2018, 47, 8572 RSC.
- M. Lomora, G. Gunkel-Grabole, S. Mantrib and C. G. Palivan, Chem. Commun., 2017, 53, 10148 RSC.
- M. R. Molla, P. Rangadurai, L. Antony, S. Swaminathan, J. J. de Pablo and S. Thayumanavan, Nat. Chem., 2018, 10, 659 CrossRef CAS.
- E. Amstad, S.-H. Kim and D. A. Weitz, Angew. Chem., Int. Ed., 2012, 124, 12667 CrossRef.
- W. Ke, J. Li, F. Mohammed, Y. Wang, K. Tou, X. Liu, P. Wen, H. Kinoh, Y. Anraku, H. Chen, K. Kataoka and Z. Ge, ACS Nano, 2019, 13, 2357 CAS.
- H. Che, S. Cao and J. C. M. van Hest, J. Am. Chem. Soc., 2018, 140, 5356 CrossRef CAS.
- R. Deng, M. J. Derry, C. J. Mable, Y. Ning and S. P. Armes, J. Am. Chem. Soc., 2017, 139, 7616 CrossRef CAS.
- S. Dhir, S. Salahub, A. S. Mathews, S. K. Kumaran, C. D. Montemagno and S. Abraham, Chem. Commun., 2018, 54, 5346 RSC.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458 CrossRef CAS.
- B. P. Bastakoti and J. Perez-Mercader, Angew. Chem., Int. Ed., 2017, 56, 12086 CrossRef CAS PubMed.
- B. P. Bastakoti and J. Perez-Mercader, Adv. Mater., 2017, 29, 1704368 CrossRef.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press/Elsevier, New York, 3rd edn, 2011 Search PubMed.
- G. Cheng and J. Pérez-Mercader, Macromol. Rapid Commun., 2018, 40, 1800513 CrossRef.
- R. Tamate, T. Ueki and R. Yoshida, Angew. Chem., Int. Ed., 2016, 55, 5179 CrossRef CAS.
- T. Litschel, B. Ramm, R. Maas, M. Heymann and P. Schwille, Angew. Chem., Int. Ed., 2018, 57, 16286 CrossRef CAS.
- R. Tamate, T. Ueki, M. Shibayama and R. Yoshida, Angew. Chem., Int. Ed., 2014, 53, 11248 CrossRef CAS.
- R. Tamate, T. Ueki and R. Yoshida, Adv. Mater., 2015, 27, 837 CrossRef CAS.
- T. Pereira de Souza and J. Perez-Mercader, Chem. Commun., 2014, 50, 8970 RSC.
- M. Orbán, K. Kurin-Csörgei and I. R. Epstein, Acc. Chem. Res., 2015, 48, 593 CrossRef.
- K. Kurin-Csörgei, I. R. Epstein and M. Orbán, Nature, 2005, 433, 139 CrossRef.
- I. Lagzi, D. Wang, B. Kowalczyk and B. A. Grzybowski, Langmuir, 2010, 26, 13770 CrossRef CAS.
- C. Zhao, J. Lu, J. Hou, X. Li, J. Wang, L. Jiang, H. Wang and H. Zhang, Adv. Funct. Mater., 2019, 29, 1806416 CrossRef.
- J. Wang, R. Fang, J. Hou, H. Zhang, Y. Tian, H. Wang and L. Jiang, ACS Nano, 2017, 11, 3022 CrossRef CAS.
- G. Wang, Y. Liu, N. Xia, W. Zhou, Q. Gao and S. Liu, Colloids Surf., A, 2017, 529, 808 CrossRef CAS.
- T. Liedl and F. C. Simmel, Nano Lett., 2005, 5, 1894 CrossRef CAS.
- A. R. Mukherjee, P. Ghosh, S. C. Chadha and S. R. Palit, Macromol. Chem. Phys., 1966, 97, 202 CrossRef CAS.
- T. G. Szántó and G. Rábai, J. Phys. Chem. A, 2005, 109, 5398 CrossRef.
- E. Poros, K. Kurin-Csörgei, I. Szalai, G. Rábai and M. Orbán, Chaos, 2015, 25, 064602 CrossRef.
- S. Svetina, ChemPhysChem, 2009, 10, 2769 CrossRef CAS.
- S. E. Maurer and G. Nguyen, Origins Life Evol. Biospheres, 2016, 46, 215 CrossRef CAS.
- L. Zhang and A. Eisenberg, Macromolecules, 1996, 29, 8805 CrossRef CAS.
- Y. Ding, M. Cai, Z. Cui, L. Huang, L. Wang, X. Lu and Y. Cai, Angew. Chem., Int. Ed., 2017, 57, 1053 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc03486b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |