
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aluminium-catalysed isocyanate trimerization, enhanced by exploiting a dynamic coordination sphere†‡
Mohammed A.
Bahili
ab,
Emily C.
Stokes
 a,
Robert C.
Amesbury
a,
Darren M. C.
Ould
a,
Bashar
Christo
a,
Rhian J.
Horne
a,
Benson M.
Kariuki
a,
Jack A.
Stewart
a,
Rebekah L.
Taylor
a,
P. Andrew
Williams
a,
Matthew D.
Jones
c,
Kenneth D. M.
Harris
a and
Benjamin D.
Ward
*a
a,
Robert C.
Amesbury
a,
Darren M. C.
Ould
a,
Bashar
Christo
a,
Rhian J.
Horne
a,
Benson M.
Kariuki
a,
Jack A.
Stewart
a,
Rebekah L.
Taylor
a,
P. Andrew
Williams
a,
Matthew D.
Jones
c,
Kenneth D. M.
Harris
a and
Benjamin D.
Ward
*a
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK. E-mail: WardBD@Cardiff.ac.uk
bDepartment of Science, College of Science, University of Basrah, Basrah, Iraq
cDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK
First published on 17th June 2019
Abstract
Main-group metals are inherently labile, hindering their use in catalysis. We exploit this lability in the synthesis of isocyanurates. For the first time we report a highly active catalyst that trimerizes alkyl, allyl and aryl isocyanates, and di-isocyanates, with low catalyst loadings under mild conditions, using a hemi-labile aluminium-pyridyl-bis(iminophenolate) complex.
Catalytic applications of Earth-abundant main-group metals, such as aluminium, have compelling environmental and economic benefits. In contrast to the extensively-used platinum group metals, they are non-toxic, inexpensive, and readily available;1 furthermore, the requirement to reprocess the metals via complex, expensive, and polluting recycling processes is therefore less critical. However, the widespread use of main group metals in catalysis is hindered by the fact that they often exhibit labile and unpredictable coordination chemistry, leading to ligand redistribution and poorly-defined catalyst species. We demonstrate here that such lability can actually be exploited as a part of the catalyst design to give a dynamic coordination environment that enhances catalytic performance.
Trimerization of isocyanates (RNCO, Scheme 1) is the most rapid, economical and atom-efficient route to isocyanurates, which are used in a diverse range of applications, such as medicines,2–5 selective anion binding,6 microporous materials7,8 and coating materials.9 However, isocyanurates’ principal application is in the construction industry; they are the key component in rigid polyurethane foams which are used ubiquitously as insulation boards and are responsible for their mechanical and thermosetting properties.10
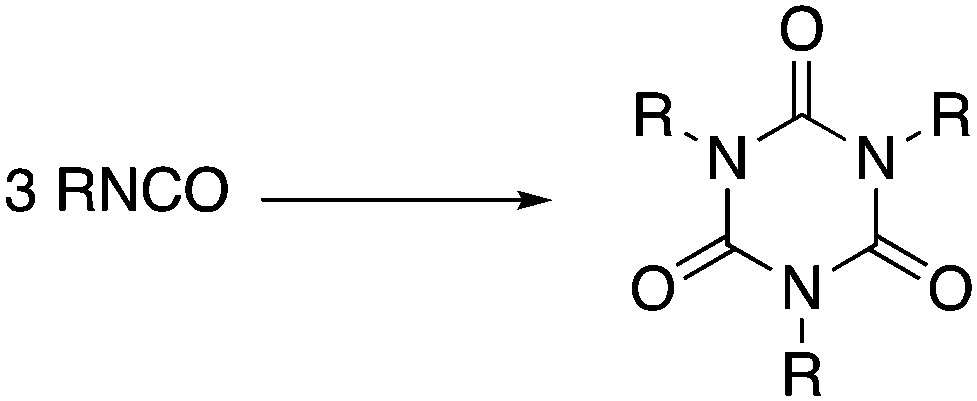 | ||
| Scheme 1 Trimerization of isocyanates. | ||
Isocyanates can be trimerized by amines,11 phosphines,12–15 N-heterocyclic carbenes,16 inorganic salts,17 phosphides,18,19 and main group and transition metal complexes,20–28 but existing catalysts often require high catalyst loadings, high temperatures (e.g. ≥100 °C for amines and halides),11,17 can give a mixture of oligomers, and/or fail to trimerize both alkyl and aryl isocyanates. In this communication we report the first catalyst that excels in all of these aspects, specifically an aluminium complex that selectively trimerizes alkyl, allyl and aryl isocyanates, and di-isocyanates, at mild temperatures, with low catalyst loadings.
Preparation of aluminium complexes. The 6-coordinate aluminium complex [Al(Salpy)(OBn)] (3) incorporating the pyridyl-bisiminophenolate (Salpy) ligand (1)29 was found to trimerize isocyanates rapidly at ambient or modest temperatures; this is remarkable since the 5-coordinate pyridyl-free analogue [Al(Salpn)(OBn)] [Salpn = bis(salicylidene)propylenediamine]30 was found to be unable to trimerize isocyanates, even under forcing conditions.§ The high activity of 3 is surprising, since a coordinatively-saturated complex is expected to be less active than a coordinatively-unsaturated one.
Reaction of the Salpy pro-ligand with AlMe3 afforded [Al(Salpy)Me] (2) in good yield (Scheme 2). Further reaction of 2 with alcohol/phenol afforded [Al(Salpy)(OR)] [R = Bn, CH2Ph, (3) or Tol, 4-C6H4Me, (4)].
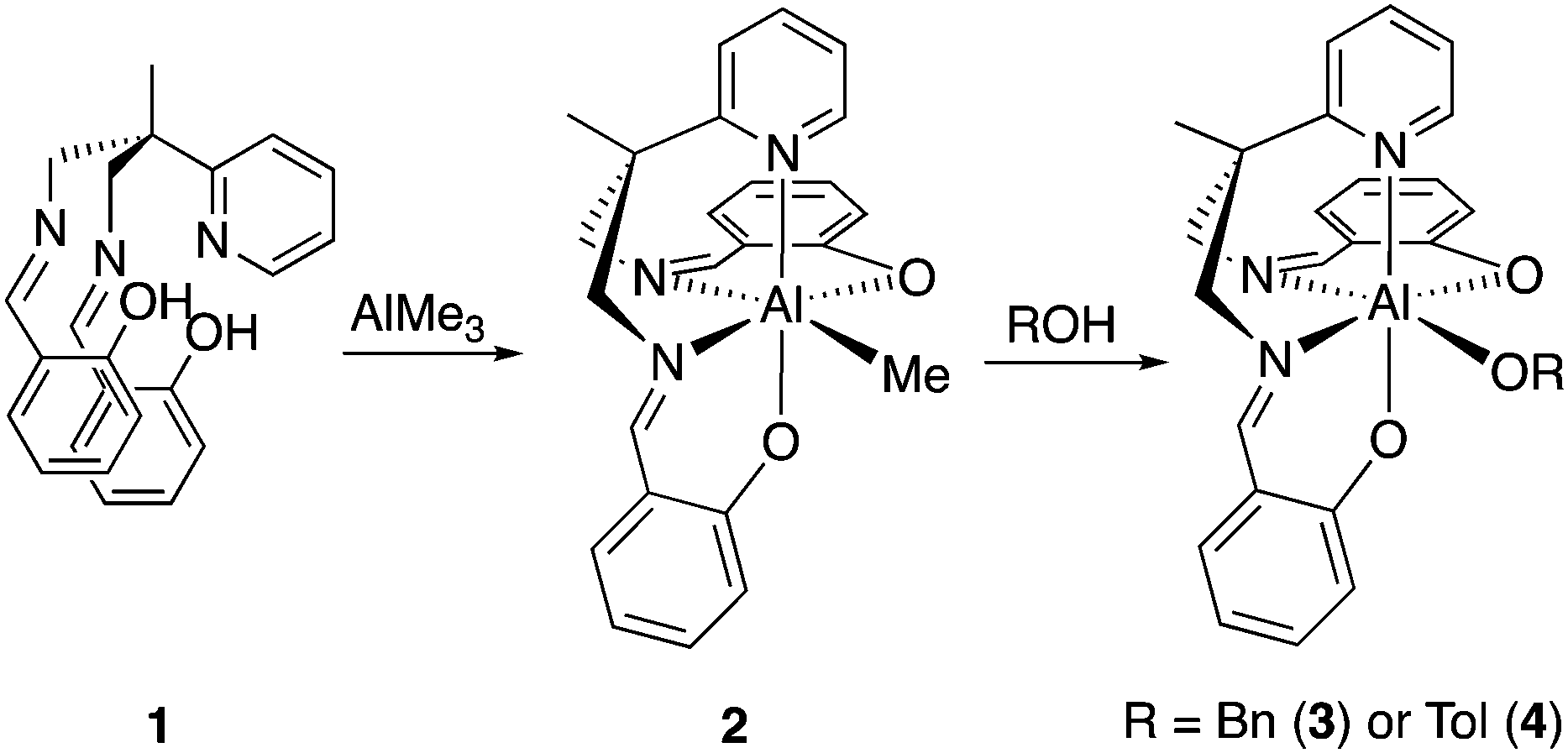 | ||
| Scheme 2 Aluminium complexes 2–4. | ||
Isocyanate trimerization. The reaction of 3 with phenyl isocyanate (PhNCO; 500 equiv.) completely solidified within 1 h at 50 °C or 18 h at 25 °C (Table 1), corresponding to essentially quantitative conversion of the isocyanate to N,N′,N′′-triphenyl isocyanurate. NMR and mass spectra indicate that the crude product mixture is pure (only trimer is detected), and the bulk product was characterized by powder X-ray diffraction (XRD). Unit cell determination and profile fitting (using the Le Bail method) led to an excellent fit to the experimental powder XRD pattern (S1.1, ESI‡). The unit cell was in excellent agreement with that of the monoclinic polymorph of the cyclotrimer reported previously.31 Rietveld refinement of the powder XRD data confirmed that the crystalline product was the monoclinic polymorph of the cyclotrimer, from which the molecular structure of the product was confirmed (see ESI‡ for more details).32
| Entry | Substrate | T (°C) | t (h) | Yieldb (%) |
|---|---|---|---|---|
| a Time taken for reaction mixture to solidify or until no further conversion attained. b Isolated yields. c Reaction carried out in the melt. | ||||
| 1 | PhNCO | 25 | 18 | 98 |
| 2 | PhNCO | 50 | 1 | 95 |
| 3 | 4-Me-C6H4NCO | 25 | 48 | 25 |
| 4 | 4-Me-C6H4NCO | 50 | 18 | 98 |
| 5 | 4-F-C6H4NCO | 25 | 6 | 93 |
| 6 | 2-F-C6H4NCO | 25 | 48 | 95 |
| 7 | 4-CF3-C6H4NCO | 50 | 18 | 95 |
| 8 | 4-Cl-C6H4NCO | 50 | 0.5 | 97 |
| 9 | 4-MeO-C6H4NCO | 50 | 3.5 | 76 |
| 10 | C6H5CH2NCO | 25 | 3 | 77 |
| 11 | H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2NCO CHCH2NCO |
25 | 3 | 59 |
| 12 | EtNCO | 25 | 0.5 | 98 |
| 13 | t BuNCO | 25 | 18 | 5 |
| 14 | t BuNCO | 50 | 18 | 96 |
| 15 | 1,4-(NCO)2C6H4 (PDI) | 100c | 1.5 | 97 |
| 16 | 1,3-(NCO)2-4-Me-C6H3 (TDI) | 25 | 1.5 | 89 |
| 17 | CH2(4-NCO-C6H4)2 (MDI) | 50c | 48 | 90 |
From our combined analyses, we conclude that the aluminium catalyst is completely selective (within detection limits) in forming the cyclotrimer, in contrast to other catalytic systems that give various side products in addition to the isocyanurate and/or require higher catalyst loadings at higher temperatures.17,33
Complex 3 is active for the trimerization of alkyl, aryl and allyl isocyanates, as well as di-isocyanates (Table 1), including those of commercial relevance (especially di-isocyanates), and those that can be incorporated into polymers as cross-linking agents (e.g. allyl isocyanate).34 From our results, it is clear that complex 3 is an exceptionally versatile catalyst, especially when considering that only low catalyst loadings are required.
Di-isocyanates find commercial use in rigid polyurethane foams and are routinely used in cavity wall, roof, and under-floor insulation. In the present work, toluene di-isocyanate (TDI), phenylene di-isocyanate (PDI) and methylenedi(phenyl-isocyanate) (MDI) were all trimerized using complex 3 (both PDI and MDI were reacted in the melt as they are solids at room temperature). For TDI and MDI, there was clear evidence (from mass spectra) for oligomeric species arising from the reaction of both isocyanate moieties, and simple trimers were also evident. The lower intensity νNCO infrared band for the product obtained from MDI compared to those for TDI indicates a greater propensity for MDI to form higher-order oligomers.
Mechanistic aspects. The structures of 2 and 4 were confirmed by single-crystal XRD (S2.3 and S2.4, ESI‡)35 and by elemental analysis and NMR spectroscopy. A second set of resonances was observed for 2 and 3 (but not for 4, suggesting that the second species is present in very low concentration). In each case, several signals due to the minor component are obscured by those of the major component, but the data are consistent with the two components being isomers; the pyridyl H6 signal for the minor isomer has a comparable chemical shift to the uncoordinated ligand (8.53 ppm for 2; 8.58 ppm for 3), while the pyridyl H6 signal for the major component is further downfield (8.69 ppm for 2 and 9.10 ppm for 3). Since the chemical shift of H6 is indicative of the coordinative state of the pyridyl,36 our observations suggest that the minor isomer has a de-coordinated pyridyl.
From 1H NMR spectra of 3 measured between 298 K and 323 K, the equilibrium constant for the major ⇌ minor component interchange was determined at each temperature and a van’t Hoff plot (S1.6, ESI‡) gave ΔH⊖ = 13.6 ± 4.2 kJ mol−1 and ΔS⊖ = 29.5 ± 13.5 J K−1 mol−1. Hence, ΔG⊖ at 298 K is estimated to be 4.8 kJ mol−1. The small value of ΔS⊖ is consistent with a unimolecular process rather than a monomer–dimer equilibrium, and the increase in entropy on going from the major to the minor component suggests that the major component has the coordinated pyridyl. The two components give almost identical diffusion coefficients in DOSY NMR spectra (S1.6 and Fig. S5, ESI‡).
The mechanism for the trimerization of MeNCO by [Al(Salpy)(OMe)] (5calc) was probed using DFT calculations. It is common for isocyanate trimerization to proceed via nucleophilic attack on the isocyanate carbon,10 and there are several examples of isolated species arising from the insertion of isocyanates into metal–ligand bonds.18,23,37–39 This leads us to propose a catalytic cycle (Scheme 3) based on a repeated coordination–insertion mechanism.¶ A crucial component of this mechanism is that pyridyl dissociation must occur before the isocyanate can associate to allow the 1,2-migratory insertion of the methoxide onto the isocyanate carbon; importantly, de-coordination of the pyridyl is facile (S to INT1: ΔG = +16 kJ mol−1). Coordination of the isocyanate to the aluminium (INT2) is endergonic (Grel = +52 kJ mol−1); INT2 lies only slightly lower in energy than the transition state TS1 and is therefore likely to be synchronous with the insertion step.
 | ||
| Scheme 3 Proposed catalytic cycle for the trimerization of MeNCO using [Al(Salpy)(OMe)] (5calc). [Al] = [Al(κ4-Salpy)]; [Al]* = [Al(κ5-Salpy)]. Gibbs energies (Grel) in kJ mol−1 calculated using M06-2X: cc-pVTZ/cc-pV(T+d)Z are shown alongside each label. The free energy profile is provided in the ESI.‡ Boxed sections highlight the pyridyl hemi-lability and its role in enhancing the catalytic activity. | ||
Bader's quantum theory of atoms in molecules analysis of INT2 indicates a bond-critical point (BCP) between the associated isocyanate carbon and the methoxide oxygen, (electron density ρ = 0.09 e Å−3) which indicates that the isocyanate is pre-organized for the migratory insertion of methoxide, i.e. the BCP lies along the reaction coordinate. All migratory insertion steps (TS1 to TS3) can be described (natural bonding orbital analyses) as a methoxide/imine sp2 hybrid donating to an empty p orbital of the isocyanate carbons (S3.2, ESI‡).
The product from the migratory insertion initially affords an O,O′-coordinated isocarbamate ligand (INT3), but as this is endergonic relative to INT2, the insertion is expected to be readily reversible without intervention of the pyridyl. However, pyridyl coordination affords a monodentate isocarbamate that has significantly lower energy (INT4, Grel = +11 kJ mol−1). Rather than re-coordination of the OMe, imine coordination is more favourable, giving OC1 which must be an off-cycle intermediate as the imine lone pair is unavailable for the subsequent migratory insertion to isocyanate. The energetics of INT4, OC1 and INT3 support a resting state pyridyl-imine coordination equilibrium between INT4 and OC1; although re-formation of INT3 is energetically viable, an INT4/OC1 equilibrium locks the isocarbamate into a configuration that conformationally disfavours OMe re-coordination by placing it away from the Al centre, reducing the likelihood of de-insertion and promoting the forward reaction, thus explaining the importance of the pyridyl donor in enhancing catalytic performance, consistent with experimental observations.
Insertion of subsequent isocyanates proceeds in a similar manner to the first. As the stepwise formation of an isocyanurate affords a linear series of amide linkages with rigid 120° bond angles, it follows that, after the third isocyanate insertion has taken place, the OMe group at the terminus is necessarily close to the imino nitrogen from the third isocyanate insertion, leading to cyclization to afford the isocyanurate and the regeneration of 5calc. For the overall reaction, ΔG is calculated to be −139 kJ mol−1.
As demonstrated in this work, the presence of a hemi-labile pyridyl donor makes the aluminium alkoxide complex [Al(Salpy)(OBn)] (3) a highly active and selective catalyst for the trimerization of isocyanates under mild conditions. Calculations suggest that the pyridyl donor facilitates the partial de-coordination of the growing trimer, thereby disfavouring the de-insertion reaction. Whilst this is an important development for isocyanurate synthesis, the concept of an additional donor group enhancing catalytic performance may have profound implications for the use of labile main group metals in a wider range of catalytic reactions, and we anticipate significant research opportunities with the aim of further exploiting this concept.
This research was supported by the Iraqi Ministry of Higher Education (studentship to MAB), the Cardiff Partnership Fund (postdoctoral fellowship to ECS), Kingspan Insulation Ltd, and the EPSRC (access to the National crystallography Service and the National Mass Spectrometry Facility, studentship to RCA, EP/L016443/1). Access to the Advanced Research Computing facility (ARCCA) at Cardiff University is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. V. Ragnarsdóttir, Nat. Geosci., 2008, 1, 720–721 CrossRef.
- P. Gibbons, D. Love, T. Craig and C. Budke, Vet. Parasitol., 2016, 218, 1–4 CrossRef CAS PubMed.
- M. Ghosh and M. J. Miller, J. Org. Chem., 1994, 59, 1020–1026 CrossRef CAS.
- A. Bosco, L. Rinaldi, G. Cappelli, A. Saratsis, L. Nisoli and G. Cringoli, Vet. Parasitol., 2015, 212, 408–410 CrossRef CAS PubMed.
- A. P. Murray and M. J. Miller, J. Org. Chem., 2003, 68, 191–194 CrossRef CAS PubMed.
- M. Mascal, I. Yakovlev, E. B. Nikitin and J. C. Fettinger, Angew. Chem., Int. Ed., 2007, 46, 8782–8784 CrossRef CAS PubMed.
- Y. Zhang, S. N. Riduan and J. Y. Ying, Chem. – Eur. J., 2009, 15, 1077–1081 CrossRef CAS PubMed.
- E. Preis, N. Schindler, S. Adrian and U. Scherf, ACS Macro Lett., 2015, 4, 1268–1272 CrossRef CAS.
- H. Ni, A. D. Skaja, R. A. Sailer and M. D. Soucek, Macromol. Chem. Phys., 2000, 201, 722–732 CrossRef CAS.
- M. F. Sonnenschein, Polyurethanes: science, technology, markets, and trends, Wiley, Hoboken, New Jersey, 2015 Search PubMed.
- Y. Taguchi, I. Shibuya, M. Yasumoto, T. Tsuchiya and K. Yonemoto, Bull. Chem. Soc. Jpn., 1990, 63, 3486–3489 CrossRef CAS.
- Z. Pusztai, G. Vlád, A. Bodor, I. T. Horváth, H. J. Laas, R. Halpaap and F. U. Richter, Angew. Chem., Int. Ed., 2006, 45, 107–110 CrossRef CAS PubMed.
- S. M. Raders and J. G. Verkade, J. Org. Chem., 2010, 75, 5308–5311 CrossRef CAS PubMed.
- J. Tang, T. Mohan and J. G. Verkade, J. Org. Chem., 1994, 59, 4931–4938 CrossRef CAS.
- X. Liu, Y. Bai and J. G. Verkade, J. Organomet. Chem., 1999, 582, 16–24 CrossRef CAS.
- H. A. Duong, M. J. Cross and J. Louie, Org. Lett., 2004, 6, 4679–4681 CrossRef CAS PubMed.
- Y. Nambu and T. Endo, J. Org. Chem., 1993, 58, 1932–1934 CrossRef CAS.
- W. Yi, J. Zhang, L. Hong, Z. Chen and X. Zhou, Organometallics, 2011, 30, 5809–5814 CrossRef CAS.
- D. Heift, Z. Benkő, H. Grützmacher, A. R. Jupp and J. M. Goicoechea, Chem. Sci., 2015, 6, 4017–4024 RSC.
- A. Hernán-Gómez, T. D. Bradley, A. R. Kennedy, Z. Livingstone, S. D. Robertson and E. Hevia, Chem. Commun., 2013, 49, 8659–8661 RSC.
- X. Zhu, J. Fan, Y. Wu, S. Wang, L. Zhang, G. Yang, Y. Wei, C. Yin, H. Zhu, S. Wu and H. Zhang, Organometallics, 2009, 28, 3882–3888 CrossRef CAS.
- Y. Wu, S. Wang, X. Zhu, G. Yang, Y. Wei, L. Zhang and H. Song, Inorg. Chem., 2008, 47, 5503–5511 CrossRef CAS PubMed.
- X. Zhou, L. Zhang, M. Zhu, R. Cai, L. Weng, Z. Huang and Q. Wu, Organometallics, 2001, 20, 5700–5706 CrossRef CAS.
- H. R. Sharpe, A. M. Geer, H. E. L. Williams, T. J. Blundell, W. Lewis, A. J. Blake and D. L. Kays, Chem. Commun., 2017, 53, 937–940 RSC.
- A. J. Bloodworth and A. G. Davies, J. Chem. Soc., 1965, 6858–6863 RSC.
- F. Moghaddam, M. Dekamin and G. Koozehgari, Lett. Org. Chem., 2005, 2, 734–738 CrossRef CAS.
- A. Flamini, A. M. Giuliani and N. Poli, Tetrahedron Lett., 1987, 28, 2169–2170 CrossRef CAS.
- S. G. Lee, K.-Y. Choi, Y.-J. Kim, S. Park and S. W. Lee, Dalton Trans., 2015, 44, 6537–6545 RSC.
- R. Shakya, A. Jozwiuk, D. R. Powell and R. P. Houser, Inorg. Chem., 2009, 48, 4083–4088 CrossRef CAS PubMed.
- H. Du, X. Pang, H. Yu, X. Zhuang, X. Chen, D. Cui, X. Wang and X. Jing, Macromolecules, 2007, 40, 1904–1913 CrossRef CAS.
- A. Usanmaz, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 1117–1119 CrossRef.
- K. D. M. Harris, Advanced X-Ray Crystallography, Springer, Berlin, Heidelberg, 2012, vol. 315, pp. 133–177 Search PubMed.
- F. Paul, S. Moulin, O. Piechaczyk, P. Le Floch and J. A. Osborn, J. Am. Chem. Soc., 2007, 129, 7294–7304 CrossRef CAS PubMed.
- W. Sun, X. Yan and X. Zhu, J. Appl. Polym. Sci., 2011, 122, 2359–2367 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- S. Friedrich, M. Schubart, L. H. Gade, I. J. Scowen, A. J. Edwards and M. McPartlin, Chem. Ber./Recl., 1997, 130, 1751–1759 CrossRef CAS.
- L. Orzechowski and S. Harder, Organometallics, 2007, 26, 2144–2148 CrossRef CAS.
- H. Wang, H.-W. Li and Z. Xie, Organometallics, 2003, 22, 4522–4531 CrossRef CAS.
- W. Uhl, J. S. Bruchhage, M. Willeke, A. Hepp and J. Kösters, Eur. J. Inorg. Chem., 2016, 2721–2730 CrossRef CAS.
Footnotes |
| † The data supporting the results presented in this article are freely available via the Cardiff University Data Catalogue at http://dx.doi.org/10.17035/d.2019.0069570379. |
| ‡ Electronic supplementary information (ESI) available: Full experimental details and characterizing data, Cartesian coordinates of all calculated species. CCDC 1896206–1896216. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc03339d |
| § Reactions with [Al(Salpn)(OBn)] were performed at 20–100 °C for 24 hours. Isocyanurate was obtained only for 4-fluorophenylisocyanate with <15% conversion, i.e. with the substrate expected to have the most electrophilic isocyanate carbon. No reaction was observed for other substrates. |
| ¶ Stoichiometric reactions on an NMR tube scale gave rise to products consistent with a sequential insertion of isocyanates, as predicted by our hypothesized mechanism. Since, the modus operandi of nucleophile-based catalysts is well established, and several isolated species reported in the literature relate to isocyanate insertion into M–L bonds, there is tangible experimental verification for the validity of the mechanism shown in Scheme 3. See ESI‡ (S1.8) for full details. |
| This journal is © The Royal Society of Chemistry 2019 |