
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism and regioselectivity of the anionic oxidative rearrangement of 1,3-diketones towards all-carbon quaternary carboxylates†
Emma
Bratt‡
a,
Samuel
Suárez-Pantiga‡
 b,
Magnus J.
Johansson
a and
Abraham
Mendoza
*b
b,
Magnus J.
Johansson
a and
Abraham
Mendoza
*b
aMedicinal Chemistry, Research and Early Development Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Pepparedsleden 1, 431 83 Mölndal, Sweden
bDept. of Organic Chemistry, Stockholm University, Arrhenius Laboratory, 106 91 Stockholm, Sweden. E-mail: abraham.mendoza@su.se
First published on 3rd June 2019
Abstract
The oxidative rearrangement of 1,3-diketones is an underexplored alternative to enolate chemistry in the synthesis of all-carbon quaternary carboxylates. The mechanistic investigation of this reaction has resulted in a mild base mediated protocol, whose regioselectivity has been studied in challenging acyclic substrates.
Carboxylic acids are widespread in natural and artificial molecules with important functions, particularly those with an all-carbon quaternary center next to the carboxylic function 1 (Scheme 1A). The synthesis of these congested products requires powerful nitrile enolates 2, 3 as masked carboxylate equivalents (Scheme 1B).1–4 However, gaining control over those processes needs cryogenic and strictly anhydrous conditions. Alternatively, the oxidative sigmatropic rearrangement of 1,3-diketones 4 offers a fundamental advantage to gain a facile entry into these compounds.5,6 The required β-diketones substrates 4 can be obtained easily through mild alkylation reactions of materials 5 with technical solvents and reagents. The formation of the key C–C bond through an intramolecular alkyl shift is advantageous over intermolecular coupling, as it is less sensitive to congested environments and can be conducted in a stereospecific fashion.7
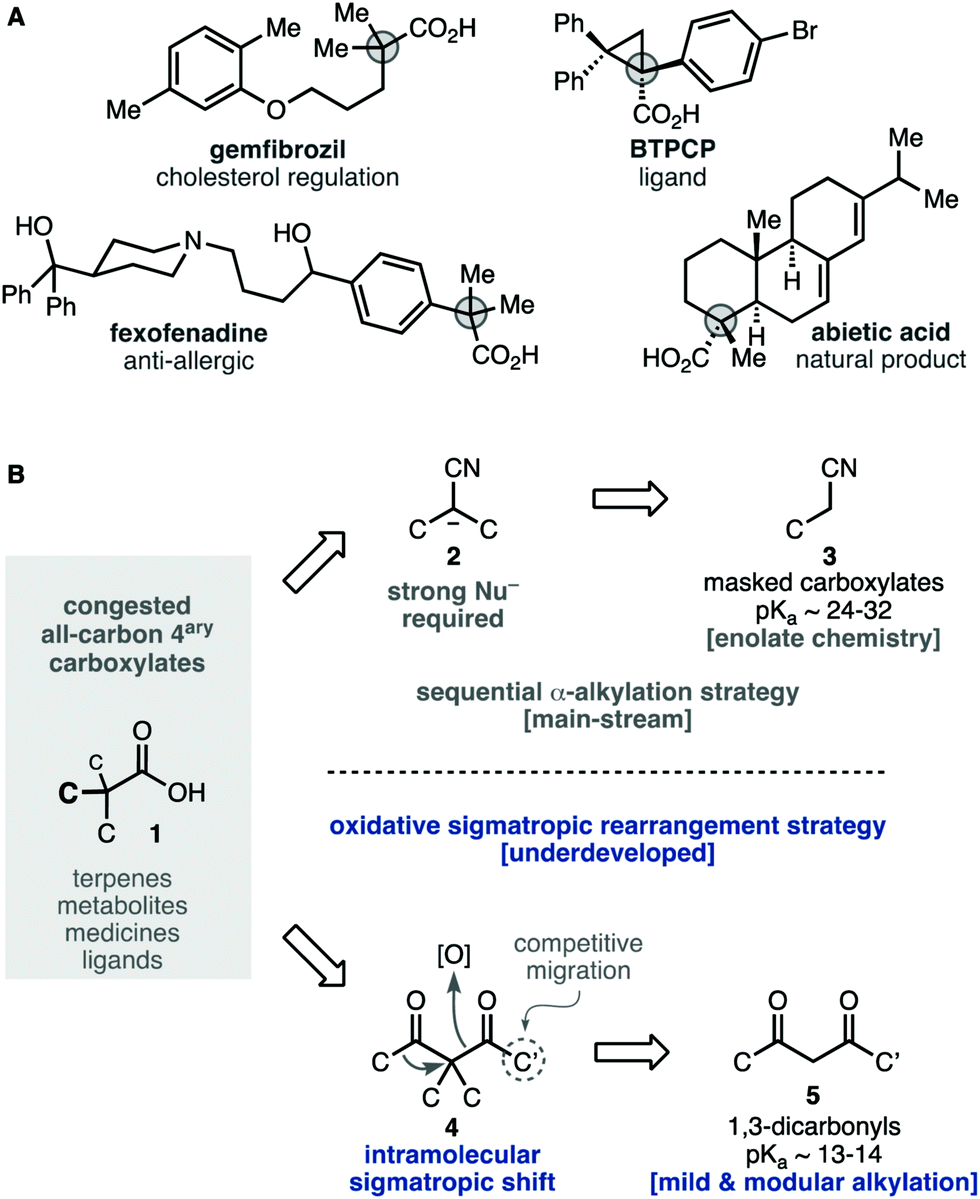 | ||
| Scheme 1 Retrosynthesis of all-carbon quaternary carboxylates and limitations of the key oxidative sigmatropic rearrangement of 1,3-diketones. | ||
However, the oxidative rearrangement of 4 has been studied and used only rarely.5 This can be explained, in part, by the substrate-dependent character of this reaction. The diketones 4a (Scheme 2A) in which one of the migrating groups is connected to the central carbon with a cyclic structure have been most successful, and produce ring-contracted carboxylic acids 1a through the endoperoxide intermediate 6a.5,7–12 In stark contrast, unbiased acyclic substrates 4b (Scheme 2B) are generally less efficient and less selective and give rise to mixtures of products 1b, b′ due to the alternative rearrangement modes of the endoperoxide intermediate 6b.7 In most reports, this rearrangement is conducted under acidic5,6,10–15 or neutral7–9,12 conditions, sometimes requiring forcing temperatures.5,6,8,14 Symmetric diketone substrates are commonplace to avoid the low regioselectivities reported in unsymmetrical diketones.7,16 As far as we are aware, the highest regioselectivity in the sigmatropic shift of carbon groups ever recorded in this reaction is 1.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.16 Recently, the study of cyclic formyl substrates revealed that the regioselectivity is highly dependent on the structure of the substrate despite the conformational bias and the stereoelectronic difference between the migrating groups.7
:1.16 Recently, the study of cyclic formyl substrates revealed that the regioselectivity is highly dependent on the structure of the substrate despite the conformational bias and the stereoelectronic difference between the migrating groups.7
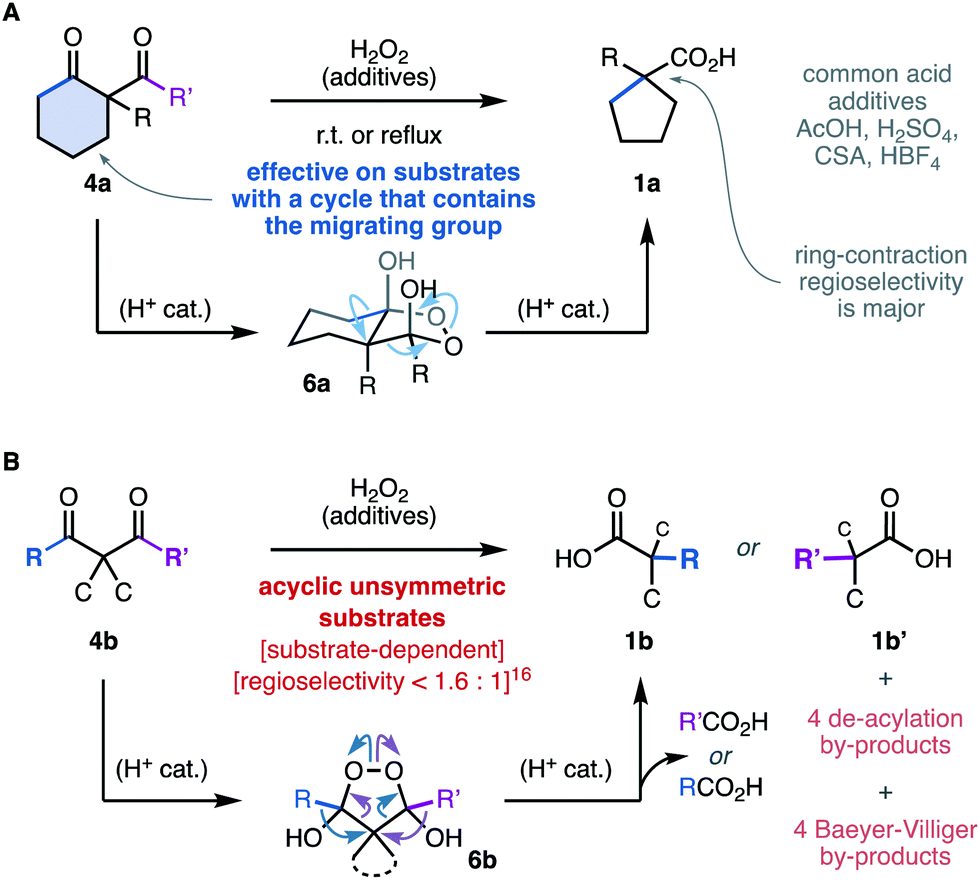 | ||
| Scheme 2 State-of-the-art in the synthesis of carboxylic acids through the oxidative rearrangement of 1,3-diketones. | ||
This reaction has been proposed to proceed through an endoperoxide dihemiketal intermediates 6.6–11,13–16 The superior performance in the presence of acidic additives5,6,10–15 was deemed to catalyse the condensation equilibrium between the diketones 4 and H2O2, which can lead to multiple adducts.12,17,18 The sigmatropic rearrangement driven by the fragmentation of the O–O bond has also been proposed to be acid catalyzed.6,16 However, there is no experimental evidence that kinetically correlate any of the possible endoperoxide intermediates12,17,18 with the formation of the carboxylic acid products 1. These fundamental challenges add to the known background reactivity of diketones 4 with H2O2 through rapid de-acylation6–9,16,17 and Baeyer–Villiger oxidation.19,20
In line with our interest in the synthesis of ligands and polar molecules,21–26 we aimed to clarify the mechanism of this reaction and the fundamental role of the various additives reported. We hoped to use this knowledge to understand the complex dynamics of this system and to control the regioselectivity of the alkyl shift in unbiased acyclic substrates. Preliminary scouting of the wide variety of conditions in literature revealed that under forcing conditions,7–9,12 the reaction was surprisingly most efficient in the absence of acetic acid (entries 1 and 2; Table 1).5,6,10–15 Milder temperature conditions were desirable to enhance the regioselectivity on unsymmetrical diketone substrates. However, only low yields were observed at room temperature (entries 3 and 4), despite the intriguing high conversion observed in neutral media (entry 3). In the presence of catalytic H2SO4, extensive decomposition occurred.
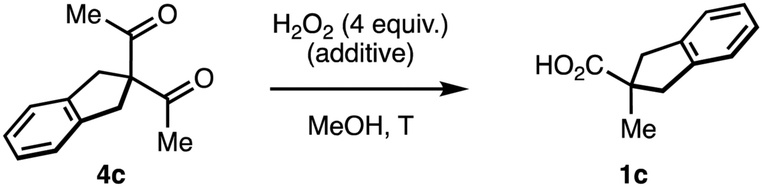
We studied the evolution of this system by in situ NMR spectroscopy.27 Taking into account the known complexity of the equilibrium between diketones and H2O2,12,17,18 the substrate 4c was deemed suitable for its symmetric structure. Particularly, the benzylic protons that do not participate in the rearrangement facilitate the monitorization (Scheme 3). Upon mixing with H2O2, we observed the slow condensation with 4c in neutral media over the course of several hours at room temperature (50% conversion at 12 hours), observing traces of the carboxylic acid product 1c. Two intermediates 6c with symmetric structure accumulated progressively over time in a 10:1 ratio. The multiplicity of their 1H-NMR signals, and additional 1D-NOE experiments further confirmed the cis relationship of the hydroxyl groups in the major diastereoisomer meso-6c, thus contradicting the previous belief that this intermediate is trans.16 The 13C-NMR spectra displayed a single signal in the acetal region (δC = 107.13 ppm), which is consistent with known endoperoxide dihemiketals (δC = 105–106 ppm),12 and allow us to discard hydroperoxyketals structures (δC ∼ 113 ppm).§12 2D-Heteronuclear correlation experiments further confirmed the connectivity of the acetal framework of the intermediate.
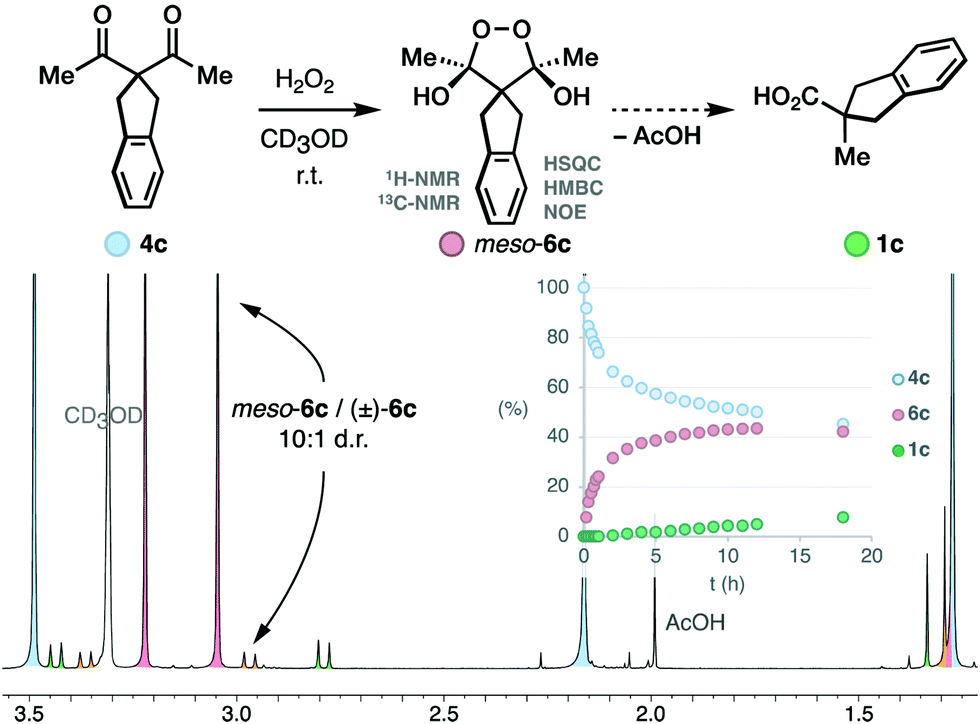 | ||
| Scheme 3 Speciation of the reaction between diketone 4c and H2O2. | ||
Brönsted acid additives are extensively used in the oxidative rearrangement of 1,3-diketones and other condensations between polyketones and H2O2.12,17,18 To our surprise, we observed complete inhibition of the reaction in the presence of acetic acid, as opposed to the slow, yet detectable, evolution in neutral media (Scheme 4A). We have confirmed that the rearrangement of an independently synthesized endoperoxide 6c into the product 1c, occurs in acidic media at a similar slow rate to that found in neutral media (see ESI†). The lack of significant acceleration in the rearrangement of endoperoxide 6c in acidic media, discards the acid-catalysis that has been assumed to operate earlier.16 Although base additives are rare in the literature of this reaction,16,28 and sometimes even declared to be detrimental to its efficiency,10 the inhibition in acidic media invited to explore their effect. Surprisingly, the presence of just 0.5 equiv. LiOH leads to a fast reaction that reaches 78% conversion in 10 minutes, and then proceeds very slowly. However, the yield of the product 1c was only 40% despite the high conversion of 4c. This seemed to point at the accumulation of intermediate 6c. Speciation of the reaction mixture over time (Scheme 4B) revealed that after the initial burst, the concentration of the endoperoxide intermediate 6c decays slowly with apparent zero-order kinetics. This occurs at a very similar rate as it does in neutral media, as evidenced by the overlay of the shifted data to account for the initial conversion that occurred in the basic reaction (Scheme 4C).
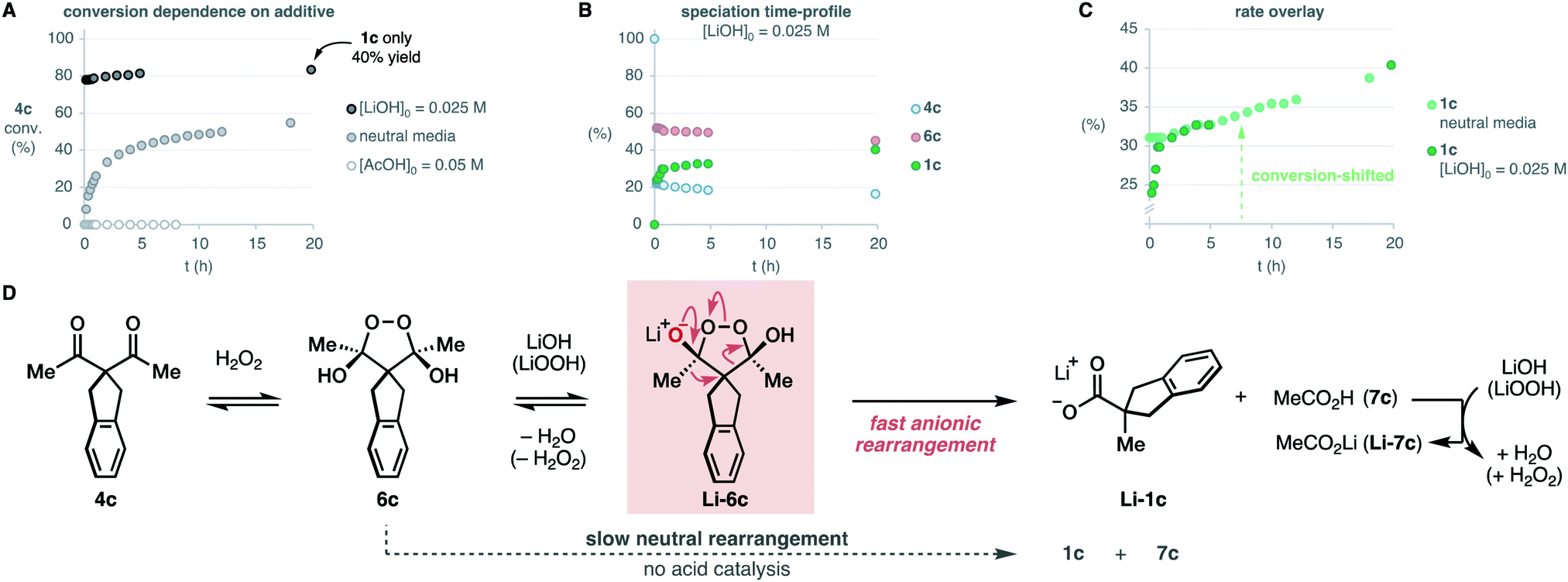 | ||
| Scheme 4 Effect of the acidic and basic additives and mechanistic proposal. Percentages are relative to the initial concentration of the limiting diketone 4c (0.05 M). | ||
All these observations point to a base-promoted sigmatropic rearrangement via intermediate Li-6c, which is self-inhibited by the formation of the carboxylic acid by-product 7c (Scheme 4D). The progress of the reaction generates carboxylic acid products that neutralize the basic media, decreasing the rate of the reaction into the apparent zero-order regime observed after the initial burst. The accumulation of the endoperoxide 6c in neutral media and its slow evolution into the carboxylate product 1c is likely due to a lower concentration of the endoperoxide anion Li-6c. As far as we know, these are the first evidences of an anionic rearrangement mechanism operating in this reaction. This mechanism does not rule out the unimolecular evolution of the endoperoxide intermediate 6c in neutral or acidic media, which is commonly proposed in the literature. However, our data strongly suggests that rearrangement from endoperoxide Li-6c is significantly faster.
In this scenario, we should expect that the rate, and most importantly, the final conversion and yield of the reaction should increase when the basicity of the reaction media is increased. We reasoned that at some point, the concentration of base should be high enough to avoid the accumulation of endoperoxide intermediate 6c, thus achieving complete conversion rapidly. To our delight, increasing amounts of LiOH enabled higher conversions and higher yields of the desired product 1c (Scheme 5A and B). In these experiments, the conversions in the initial burst correlated with the initial concentration of base. The following slow regimes had apparent zero-order with similar rates, as evidenced by the overlay of the conversion-shifted plots (Scheme 5C). This is consistent with the expected self-neutralization of the reaction medium at different conversions, depending on the initial concentration of base (vide supra). With 4 equivalents of LiOH we only transiently detected the endoperoxide intermediate 6c, thus leading to high yields of 1c rapidly (Scheme 5D). The reaction was equally efficient with NaOH or KOH, which indicates that lithium does not play a major role in the rearrangement (see ESI†). Pre-mixing of base and H2O2 generated the milder base LiOOH in situ, which resulted in slightly faster conversion (see ESI†). Although we never experienced any safety issues, the diketone was added at 0 °C to control possible exotherms. Upon warming to room temperature, we obtained the desired product 1c in 96% yield in less than 10 minutes, as evidenced by 1H-NMR.
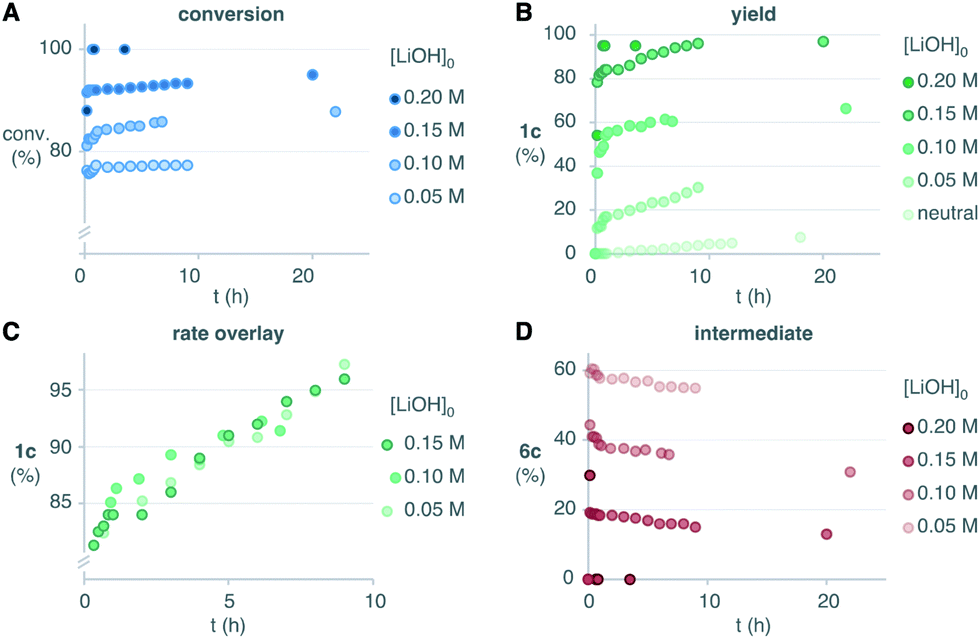 | ||
| Scheme 5 Effect of the initial concentration of base additive in the conversion, yield and accumulation of endoperoxide intermediate. Percentages are relative to the initial concentration of the limiting diketone 4c (0.05 M). | ||
The mild protocol developed with the guidance of intermediate speciation, allowed to explore the intrinsic regioselectivity of the sigmatropic rearrangement. We set out to compare the migrating ability of different carbon groups, relative to that of methyl, using model substrates 4c–k. Primary and secondary alkyls, including cyclopropyl and benzyl, where deemed suitable for the rearrangement, albeit with poor regio-discrimination (entries 1–5, Table 2). Bearing in mind that Li-6 is reminiscent of a semi-pinacol intermediate, it was expected that substituents with higher s-character would be more easily outcompeted by alkyl groups. Indeed, aryl-substituents significantly increased the selectivity, while preserving the overall efficiency of the process (entries 6 and 7). The selectivity was increased further using electron deficient aromatics, likely due to the decreased electron density in their shifting carbon (entries 8 and 9). This effect was particularly noteworthy in the case of the p-trifluorotolyl derivative 4k, in which the rearrangement proceeded with 31:1 selectivity (entry 9), thus surpassing the highest 1.6:1 regioselectivity reported between carbon substituents.16 While slow aryl migration benefits the regioselective alkyl shift studied herein, further developments are required to invert this trend to access interesting α-arylcarboxylates (Scheme 1A).
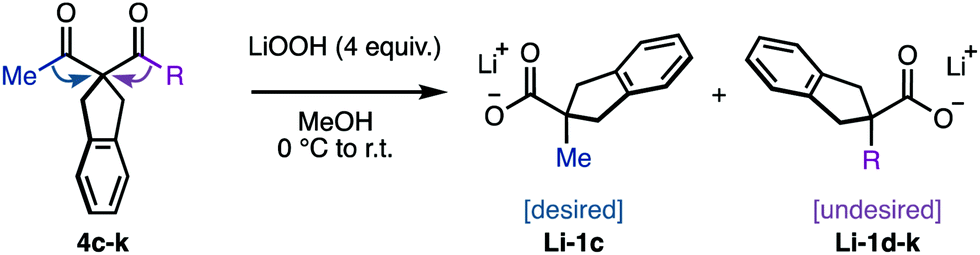
To summarize, the characterization and speciation of intermediates in the oxidative rearrangement of 1,3-diketones allowed to rationalize the efficiency of a mild, efficient and practical protocol in basic media. The identification of the self-inhibition profile of this reaction proved key to optimize the current conditions. The stereochemistry of the intermediate endoperoxide dihemiketal intermediate, and its competence relative to its anionic derivative, challenge previous proposed mechanisms. The mild conditions of the rearrangement promoted by LiOOH allowed to explore the relative intrinsic regioselectivity of the migration and allowed the rational design of a promising spectator group. These discoveries enable future applications of this reaction in the synthesis of complex molecules with valuable migrating groups, which we are currently exploring.
We thank Jorge Otero and Mulima Mabbanti for their contributions, and AstraZeneca Gothenburg and Stockholm University for unrestricted support. We are grateful to the Wallenberg Foundation (KAW2016.0153), and the ERC (714737). NMR raw data is available DOI: http://10.5281/zenodo.3234375.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. A. S. Smith, G. J. W. Breen, M. K. Hajek and D. V. C. Awang, J. Org. Chem., 1970, 35, 2215 CrossRef CAS.
- B. Corbel and T. Durst, J. Org. Chem., 1976, 41, 3648 CrossRef CAS.
- D. Savoia, E. Tagliavini, C. Trombini and A. Umani-Ronchi, J. Org. Chem., 1980, 45, 3227 CrossRef CAS.
- C. G. Espino, K. W. Fiori, M. Kim and B. Du, J. Am. Chem. Soc., 2004, 126, 15378 CrossRef CAS PubMed.
- C. Mannich, Chem. Ber., 1941, 74, 1007 CrossRef.
- G. Payne, J. Org. Chem., 1961, 26, 4793 CrossRef CAS.
- X. Yu, J. Hu, Z. Shen, H. Zhang, J.-M. Gao and W. Xie, Angew. Chem., Int. Ed., 2017, 56, 350 CrossRef CAS PubMed.
- S. I. Zavialov, L. P. Vinogradova and G. V. Kondratieva, Tetrahedron, 1964, 20, 2745 CrossRef.
- E. G. E. Hawkins and R. Large, J. Chem. Soc., Perkin Trans. 1, 1973, 2169 RSC.
- S. Challenger, A. Derrick and T. V. Silk, Synth. Commun., 2002, 32, 2911 CrossRef CAS.
- S. Challenger, A. Derrick and T. V. Silk, Synth. Commun., 2006, 32, 2911 CrossRef.
- V. L. Novikov and O. P. Shestak, Russ. Chem. Bull., 2013, 62, 2171–2190 CrossRef CAS.
- G. Payne, J. Org. Chem., 1959, 24, 1830 CrossRef CAS.
- A. Rieche and C. Bischoff, Chem. Ber., 1963, 96, 2607 CrossRef CAS.
- X. Yu, Z. Liu, Z. Xia, Z. Shen, X. Pan, H. Zhang and W. Xie, RSC Adv., 2014, 4, 53397 RSC.
- W. Cocker and D. H. Grayson, J. Chem. Soc., Perkin Trans. 1, 1975, 1347 RSC.
- A. O. Terent'ev, D. A. Borisov, V. V. Chernyshev and G. I. Nikishin, J. Org. Chem., 2009, 74, 3335 CrossRef PubMed.
- A. O. Terent'ev, I. A. Yaremenko, V. V. Chernyshev, V. M. Dembitsky and G. I. Nikishin, J. Org. Chem., 2012, 77, 1833 CrossRef PubMed.
- H. O. House and W. F. Gannon, J. Org. Chem., 1958, 23, 879 CrossRef CAS.
- J. Cossy, B. Gille and V. Bellosta, Tetrahedron Lett., 1998, 39, 4459 CrossRef CAS.
- S. Suárez-Pantiga, K. Colas, M. J. Johansson and A. Mendoza, Angew. Chem., Int. Ed., 2015, 54, 14094 CrossRef PubMed.
- A. Mendoza, K. Colas, S. Suárez-Pantiga, D. C. G. Götz and M. J. Johansson, Synlett, 2016, 1753 CrossRef CAS.
- K. Colas, R. Martín-Montero and A. Mendoza, Angew. Chem., Int. Ed., 2017, 56, 16042 CrossRef CAS PubMed.
- J. Otero-Fraga, M. Montesinos-Magraner and A. Mendoza, Synthesis, 2017, 802 CAS.
- J. Otero-Fraga, S. Suárez-Pantiga, M. Montesinos-Magraner, D. Rhein and A. Mendoza, Angew. Chem., Int. Ed., 2017, 56, 12962 CrossRef CAS PubMed.
- M. Montesinos-Magraner, M. Costantini, R. Ramírez-Contreras, M. E. Muratore, M. J. Johansson and A. Mendoza, Angew. Chem., Int. Ed., 2019, 58, 5930 CrossRef CAS PubMed.
- (a) S. Wang, N. Lokesh, J. Hioe, R. M. Gschwind and B. König, Chem. Sci., 2019, 10, 4580 RSC; (b) C. P. Johnston, T. H. West, R. E. Dooley, M. Reid, A. B. Jones, E. J. King, A. G. Leach and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2018, 140, 11112 CrossRef CAS PubMed; (c) S. Aikonen, M. Muuronen, T. Wirtanen, S. Heikkinen, J. Musgreave, J. Burés and J. Helaja, ACS Catal., 2018, 8, 960 CrossRef CAS; (d) X. Companyó and J. Burés, J. Am. Chem. Soc., 2017, 139, 8432 CrossRef PubMed; (e) H. Bartling, A. Eisenhofer, B. König and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 11860 CrossRef CAS PubMed; (f) J. Burés, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 6741 CrossRef PubMed.
- T.-L. Ho, Synth. Commun., 2006, 13, 761 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc03331a |
| ‡ These authors contributed equally to this work. |
| § Fast fragmentation prevented HRMS analysis. |
| This journal is © The Royal Society of Chemistry 2019 |