
[2+2] Halogen-bonded boxes employing azobenzenes†
Esther
Nieland
 ,
Thomas
Topornicki
,
Tom
Kunde
and
Bernd M.
Schmidt
*
,
Thomas
Topornicki
,
Tom
Kunde
and
Bernd M.
Schmidt
*
Institut für Organische Chemie und Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, D-40225 Düsseldorf, Germany. E-mail: Bernd.Schmidt@hhu.de
First published on 7th May 2019
Abstract
Herein, we report the synthesis and crystal structures of three [2+2] supramolecular boxes assembled by halogen bonding. The discrete, two-dimensional boxes with a length of 25–30 Å are based on rigid u-shaped halogen acceptors paired with highly fluorinated, azobenzenes bearing halogen bond donors.
In principal, different intermolecular interactions have specific characteristics, such as directionality, strength, selectivity and tuneability.1 The halogen bond is an attractive non-covalent interaction between a polarized halogen atom (donor) and a Lewis base (acceptor).2 Neutral halogen bonds can be described by R–X⋯Y, where X is the halogen bond donor, R is covalently bound to X, and Y is the Lewis basic halogen bond acceptor.3 Halogen bonding can be very effectively used to assemble molecules into supramolecular architectures4 as well as discrete supermolecules5 because of its high directionality and favourable bond strength, allowing for both error correction but also for the isolation of the desired target compounds.4 Halogen bonding was successfully used to construct three-dimensional supramolecular networks4e–j and halogen-bonded liquid crystals4d,e that show enhanced stability and enable control over mesophase formation by varying the degree of fluorination accompanied by change of the halogen bond's strength.4e A prominent halogen-bonded container molecule was prepared by Diederich and co-workers5d and thoroughly investigated in the solid state, in solution and in the gas phase.5b Triangular macrocycles assembled by self-complemented halogen bonding have been reported as well.5a Additionally, several charged supramolecular capsules, including a I12L6-type halogen-bonded hexameric capsule with a diameter of 2.5 nm,6a have been reported employing halonium ions [N⋯I+⋯N], which are able to form three-centre-four-electron bonds with two halogen bond acceptor moieties and can be synthesized by cation exchange from metallosupramolecular Ag+ capsules.6
Azobenzenes in general offer reliable and clean photoreactions (minimal photo-bleaching)7 with a pronounced change in the geometry upon E to Z isomerization making them also suitable synthons for supramolecular systems.8 Especially fluorinated azobenzenes7 quickly became an important element in the creation of optical or photoresponsive materials by co-crystal formation9 and show photomechanical effects in the crystalline state.9e Azobenzenes functionalized by ortho-fluorine substituents allow selective addressing of both isomers in the visible region of the spectrum, accompanied by long thermal half-lives.7
Herein, we report the synthesis of halogen-bonded, discrete [2+2] supramolecular boxes. The boxes are based on rigid u-shaped anthracene and anthracene-9,10-dione building blocks bearing two 3,5-lutidine acceptors in 1 and 8 position combined with E-4,4′-di(iodo)perfluoroazobenzene (A1) and novel E-4,4′-di(iodoethynyl)perfluoroazobenzene (A2) acceptors (Scheme 1). Solvent-free single-crystals of U1⋯A1 and U2⋯A1, as well as a chloroform adduct of U1⋯A2 could be obtained and the solid-state structures were studied by single-crystal X-ray analysis.
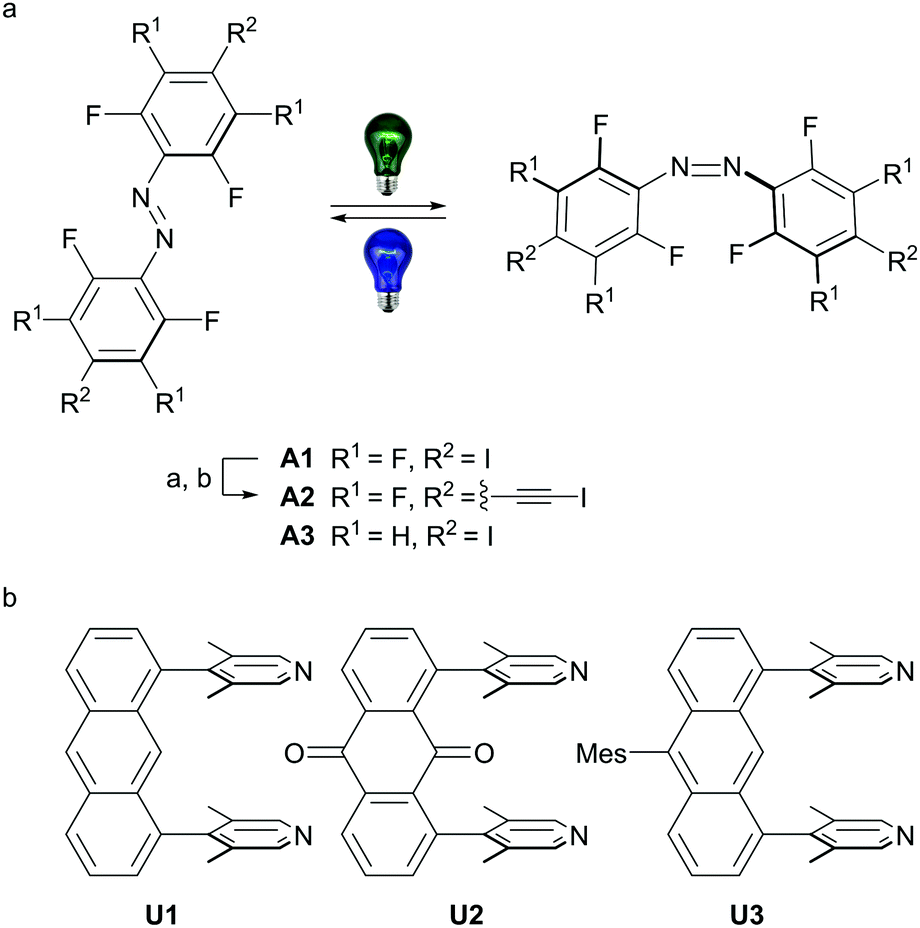 | ||
| Scheme 1 (a) Azobenzenes employed in this study, A1 was synthesized according to literature procedure starting from 2,3,5,6-tetrafluoroaniline via 4-iodo-2,3,5,6-tetrafluoroaniline.9eA2 was synthesized from A1: (a) 20.0 eq. trimethylsilylacetylene, 0.10 eq. CuI, 4.0 mol% Pd(PPh3)Cl2, triethylamine, 90 °C, 5 h, followed by (b) 2.2 eq. AgF, 2.2 eq. NIS, acetonitrile, RT, 3 h; A3 was synthesized from 2,6-difluoro-4-iodoaniline.7b (b) Synthesis of the u-shaped acceptors U1–U3 was accomplished starting from 1,8-dichloroanthraquinone (see ESI† for all detailed synthetic procedures). Mes = 1,3,5-trimethylbenzene. | ||
Equimolar amounts of U1 & A1 were dissolved in acetonitrile furnishing red crystals in quantitative yield after slow evaporation at room temperature. Single-crystal analysis confirms the formation of U1⋯A1 box in the solid state (Fig. 1).
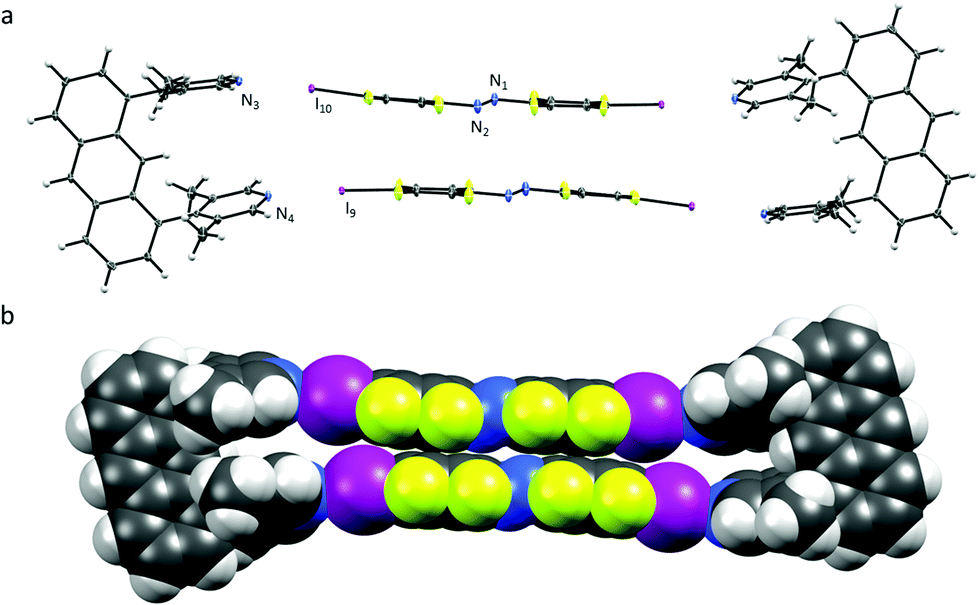 | ||
| Fig. 1 (a) X-ray crystal structure of U1⋯A1, thermal ellipsoids drawn at 50% probability level; (b) space-filling model of U1⋯A1. | ||
The U1⋯A1 box (Fig. 1) has a length of approximately 25 Å (anthracene–anthrance distance) and a height of 5 Å (distance between the ipso-carbons of the lutidines). The lutidine units are curved inwards slightly, (N⋯I–C angles vary from 172–178°) possibly to maximize the short halogen bonding interactions (N⋯I distances 2.76 and 2.78 Å) between the stacking azobenzenes A1. Azobenzenes A1 interact closely by offset π⋯π stacking (3.68 Å centroid-to-centroid distance of the benzenes) and N⋯N contacts (2.98 Å), whereas the anthracenes are engaging in an overall herringbone type arrangement (Fig. S7, ESI†) supported by C–H⋯π and C–H⋯F contacts.10 The U1⋯A2 box (Fig. 2) is consequently elongated to a length of 30 Å while maintaining a height of 5 Å. Due to the low solubility of A2 in acetonitrile, orange crystals were grown by slow evaporation of a chloroform solution containing equimolar amounts of U1 and A2.
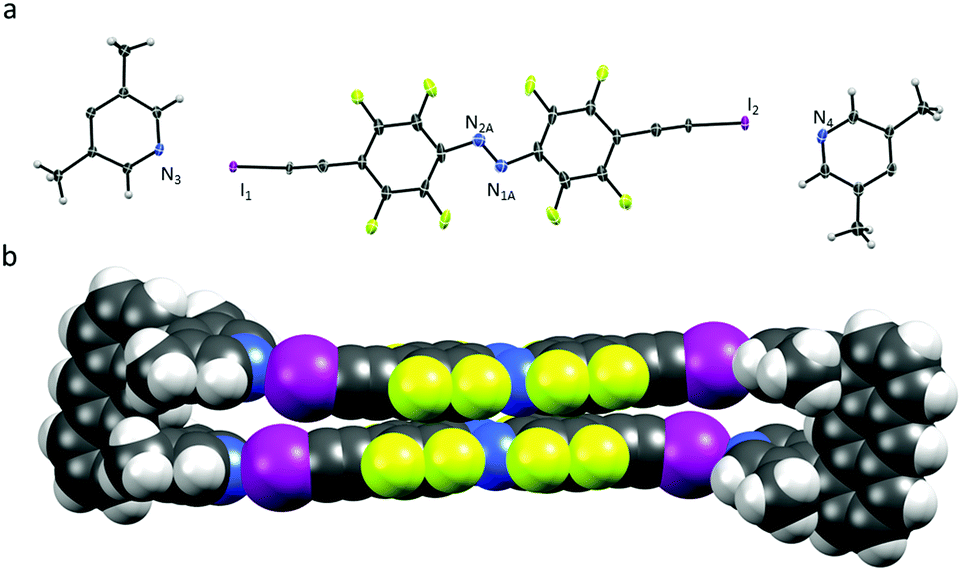 | ||
| Fig. 2 (a) Part of the X-ray crystal structure of U1⋯A2 showing the kinked alignment with thermal ellipsoids drawn at 50% probability level. The structure was solved by using a disorder model for the azo-bond of azobenzene A2, which adopts two different orientations within the crystal lattice. (b) Space-filling model of U1⋯A2. Chloroform molecules and azobenzene disorder omitted for clarity. | ||
Azobenzenes A2 interact even closer by offset π⋯π stacking (3.62 Å) and N⋯I distances of 2.76 and 2.78 Å, similar to the ones observed in U1⋯A1. The ordered chloroform molecule is located in close proximity to the azobenzene A2 but does not seem to interact with halogen bond acceptor lutidines. Short contacts are only observed between a methyl group of a neighbouring molecule (Cl⋯H 2.88 Å). Surprisingly U1⋯A2 is slightly kinked with N⋯I–C angles varying from 167–168°, deviating significantly from linearity (Fig. 2). Illumination of both single-crystals with a green LED (565 nm) resulted in no measurable isomerization of A1 or A2 in the solid state.
In contrast to U1⋯A1, the azobenzenes A1 in U2⋯A1 are rotated (Fig. 3.). The angle between the planes formed by one of the six-membered rings of the azobenzene and the lutidine is close to 81°, the halogen-bonding donor and acceptor units are almost perpendicular to each other. N⋯I–C angles vary from 169–173° with N⋯I distances similar to U1⋯A1 of 2.76 and 2.78 Å. Each of the azobenzenes A1 stacks offset not with an A1 of the same box, but with azobenzene A1 of a neighbouring box (π⋯π stacking 3.90 Å centroid-to-centroid distance of the benzenes, Fig. S11–S13, ESI†) together with short N⋯N contacts (2.98 Å). The volume of the box is much larger as the lutidine groups are pointing outwards, possibly to avoid repulsion between the ketone's lone pairs and the π-system. Attempts to crystallize U2⋯A3 did not lead to co-crystal formation but to individual single-crystals of U2 and A3 from acetonitrile solution (Fig. S14 and S15, ESI†).11
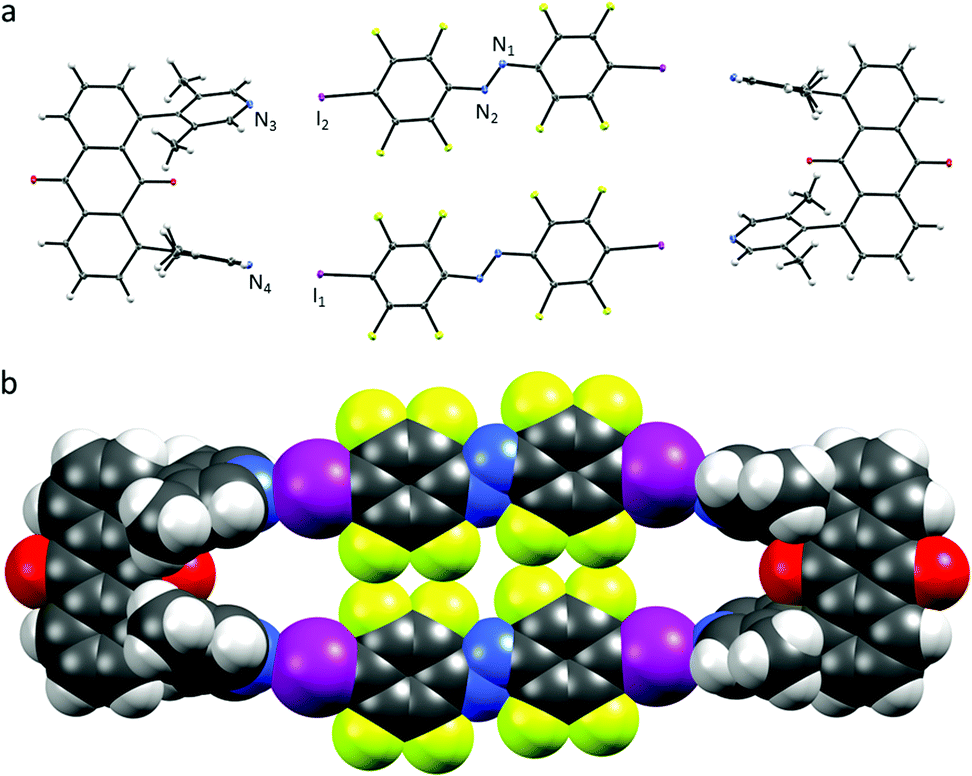 | ||
| Fig. 3 (a) X-ray crystal structure of U2⋯A1, thermal ellipsoids drawn at 50% probability level; (b) space-filling model of U2⋯A1. | ||
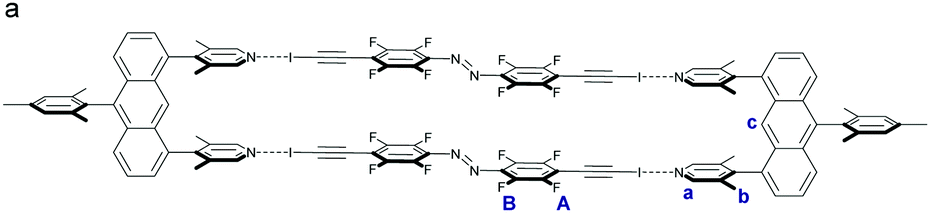
The first attempts to analyse the formation of U2⋯A1 in solution using benzene led to immediate precipitation of U2⋯A1 as an orange powder. Therefore, a mixture of chloroform and benzene (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) was used to solubilize the U2⋯A1 box. We expected to see significant upfield shifts in the 19F NMR for the fluorine atoms FA and a downfield shift for the meta positioned FB atoms, however no significant shift could be observed, regardless of the concentration.
:7) was used to solubilize the U2⋯A1 box. We expected to see significant upfield shifts in the 19F NMR for the fluorine atoms FA and a downfield shift for the meta positioned FB atoms, however no significant shift could be observed, regardless of the concentration.
To be able to observe the formation of the halogen-bonded boxes in solution we optimized the system as well as the experimental parameters. The temperature for our binding studies was lowered to 283 K in order to minimize unfavoured TΔSXB term influence upon association, as also previously described for the system of Diederich and co-workers5b,d and to minimize solvent effects, non-polar d6-benzene was used.12 The solubility of the assemblies in benzene was improved by adding a solubilizing mesitylene group to the halogen bonding acceptor (U3) to avoid precipitation5d of box U3⋯A2 during titration experiments. The halogen bond donor moiety was also strengthened by introduction of two iodoethynyl groups (A2).5b
Finally, we could confirm the 2:2 stoichiometry of U3⋯A2 (Scheme 2) by the method of continuous variation (Job plot analysis) in solution (Fig. S1, ESI†). Association constants were determined to be Ka < 102 M−1 and are on the lower detection limit with errors conservatively estimated to be in the same magnitude as the actual binding constant (Fig. S3 and S4, ESI†). The strongest change in chemical shifts for U3⋯A2 was observed to be [Δδmax(U3, Ha) = 0.05 ppm] under stated conditions. However, we did not expect the association constant to be that low because of competition, since we did not use a mixed solvent system, but due to unfavourable thermodynamics, in specific a low binding enthalpy. Under the mentioned conditions, a change in the chemical shift upon titration with A1 was observed for U3⋯A1 as well (but smaller than that of A2 in solution, (Fig. S2, ESI†)). Iodoethynyl E-A2 undergoes clean photoreaction in acetonitrile (Fig. S16, ESI†) and switches when bound as U3⋯A2 in benzene solution to Z-A2, when irradiated by a green light LED, with a photostationary state greater than 70% as determined from 19F NMR integration.
 | ||
| Scheme 2 (a) Schematic representation of U3⋯A2 box assembled from four parts as observed in the crystal structure (Fig. 3). | ||
Our results not only show that halogen bonding can be used to design highly defined non-covalent assemblies of various sizes in solution and in the solid state. To the best of our knowledge, this system is the first discrete [2+2] supramolecular box assembled by halogen bonding allowing in principle for reversible photochemical control of the geometry. Although precise NMR analysis was not possible, the change of the geometry of azobenzenes upon switching must necessarily be followed by an overall change of the geometry of the halogen bonded systems. Only few discrete halogen-bonded adducts5 can be found in the literature and the principles disclosed here may pave the way towards the design and realization of more complex and potentially responsive, halogen-bonded supramolecular systems. The ability of the supramolecular boxes described here to possibly host flat aromatic compounds with complimentary quadrupole moment is under investigation.
This work was supported by the Fonds der Chemischen Industrie via a Kekulé Fellowship (T. K.) and the Strategic Research Fund of Heinrich Heine University (F-2018/1460-4). We thank C. Czekelius, F. Q. Römpp and the NMR service team (M. Beuer and K. Schaper) of the Department of Chemistry of Heinrich Heine University Düsseldorf.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310 CrossRef CAS PubMed and references cited herein; (b) M. D. Perera, J. Desper, A. S. Sinha and C. B. Aakeröy, CrystEngComm, 2016, 18, 8631 RSC.
- (a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478 CrossRef CAS; (b) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118 CrossRef CAS.
- (a) C. B. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem. – Eur. J., 2013, 19, 16240 CrossRef PubMed; (b) G. Resnati, E. Boldyreva, P. Bombicz and M. Kawano, IUCrJ, 2015, 2, 675 CrossRef CAS.
- (a) V. Dichiarante, T. Kaio, P. Metrangolo, T. Pilati, G. Resnati and M. Ursini, Chem. Commun., 2019, 55, 4234 RSC; (b) P. M. J. Szell, S. Zablotny and D. L. Bryce, Nat. Commun., 2019, 10, 916 CrossRef PubMed; (c) M. Saccone, M. Spengler, M. Pfletscher, K. Kuntze, M. Virkki, C. Wölper, R. Gehrke, G. Jansen, P. Metrangolo, A. Priimagi and M. Giese, Chem. Mater., 2019, 31, 462 CrossRef CAS; (d) M. Saccone, K. Kuntze, Z. Ahmed, A. Siiskonen, M. Giese and A. Priimagi, J. Mater. Chem. C, 2018, 6, 9958 RSC; (e) C. Han, D. Zhao and S. Dong, Chem. Commun., 2018, 54, 13099 RSC; (f) V. I. Nikolayenko, D. C. Castell, D. P. van Heerden and L. J. Barbour, Angew. Chem., Int. Ed., 2018, 57, 12086 CrossRef CAS PubMed; (g) M. C. Pfrunder, A. J. Brock, J. J. Brown, A. Grosjean, J. Ward, J. C. McMurtrie and J. K. Clegg, Chem. Commun., 2018, 54, 3974 RSC; (h) P. Liu, Z. Li, B. Shi, J. Liu, H. Zhu and F. Huang, Chem. – Eur. J., 2018, 24, 4264 CrossRef CAS; (i) L. Bai, P. Bose, Q. Gao, Y. Li, R. Ganguly and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 436 CrossRef CAS PubMed; (j) A. Priimagi, G. Cavallo, A. Forni, M. Gorynsztejn-Leben, M. Kaivola, P. Metrangolo, R. Milani, A. Shishido, T. Pilati, G. Resnati and G. Terraneo, Adv. Funct. Mater., 2012, 22, 2572 CrossRef CAS.
- (a) P. M. J. Szell, A. Siiskonen, L. Catalano, G. Cavallo, G. Terraneo, A. Priimagi, D. L. Bryce and P. Metrangolo, New J. Chem., 2018, 42, 10467 RSC; (b) O. Dumele, B. Schreib, U. Warzok, N. Trapp, C. A. Schalley and F. Diederich, Angew. Chem., Int. Ed., 2017, 56, 1152 CrossRef CAS PubMed; (c) M. A. Sinnwell and L. R. MacGillivray, Angew. Chem., Int. Ed., 2016, 55, 3477 CrossRef CAS PubMed; (d) O. Dumele, N. Trapp and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 12339 CrossRef CAS; (e) S. H. Jungbauer, D. Bulfield, F. Kniep, C. W. Lehmann, E. Herdtweck and S. M. Huber, J. Am. Chem. Soc., 2014, 136, 16740 CrossRef CAS PubMed.
- (a) L. Turunen, U. Warzok, C. A. Schalley and K. Rissanen, Chem, 2017, 3, 861 CrossRef CAS; (b) U. Warzok, M. Marianski, W. Hoffmann, L. Turunen, K. Rissanen, K. Pagel and C. A. Schalley, Chem. Sci., 2018, 9, 8343 RSC; (c) L. Turunen, A. Peuronen, S. Forsblom, E. Kalenius, M. Lahtinen and K. Rissanen, Chem. – Eur. J., 2017, 23, 11714 CrossRef CAS; (d) L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley and K. Rissanen, Angew. Chem., Int. Ed., 2016, 55, 14033 CrossRef CAS.
- (a) C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bléger, Chem. – Eur. J., 2014, 20, 16492 CrossRef CAS PubMed; (b) D. Bléger, J. Schwarz, A. M. Brouwer and S. Hecht, J. Am. Chem. Soc., 2012, 134, 20597 CrossRef PubMed.
- (a) M. Mauro, J. Mater. Chem. B, 2019 10.1039/C8TB01893F; (b) J. Vapaavuori, C. G. Bazuin and A. Priimagi, J. Mater. Chem. C, 2018, 6, 2168 RSC; (c) L. Wang and Q. Li, Chem. Soc. Rev., 2018, 47, 1044 RSC; (d) S. Yagai, T. Karatsu and A. Kitamura, Chem. – Eur. J., 2005, 11, 4054 CrossRef CAS PubMed.
- (a) J.-C. Christopherson, F. Topić, C. J. Barrett and T. Friščić, Cryst. Growth Des., 2018, 18, 1245 CrossRef CAS; (b) O. S. Bushuyev, T. Friščić and C. J. Barrett, Cryst. Growth Des., 2016, 16, 541 CrossRef CAS; (c) O. S. Bushuyev, D. Tan, C. J. Barrett and T. Friščić, CrystEngComm, 2015, 17, 73 RSC; (d) M. Saccone, G. Terraneo, T. Pilati, G. Cavallo, A. Priimagi, P. Metrangolo and G. Resnati, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 149 CrossRef CAS PubMed; (e) O. S. Bushuyev, A. Tomberg, T. Friščić and C. J. Barrett, J. Am. Chem. Soc., 2013, 135, 12556 CrossRef CAS PubMed.
- (a) M. Nishio, CrystEngComm, 2004, 6, 130 RSC; (b) R. Fröhlich, T. C. Rosen, O. G. J. Meyer, K. Rissanen and G. Haufe, J. Mol. Struct., 2006, 787, 50 CrossRef; (c) T. S. Thakur, M. T. Kirchner, D. Bläser, R. Boese and G. R. Desiraju, CrystEngComm, 2010, 12, 2079 RSC; (d) D. Chopra and T. N. Guru Row, CrystEngComm, 2011, 13, 2175 RSC; (e) B. M. Schmidt, A. K. Meyer and D. Lentz, CrystEngComm, 2017, 19, 1328 RSC; (f) I. Birkenfelder, J. Gurke, L. Grubert, S. Hecht and B. M. Schmidt, Chem. – Asian J., 2017, 12, 3156 CrossRef CAS.
- We also prepared E-4,4′-di(iodo)-2,6-difluoroazobenzene A3 in order to be able to measure 1H DOSY NMR with 1H “handles” on halogen bond acceptor and donor but did not observe association in solution. 19F DOSY NMR cannot be routinely measured easily, see (a) J. E. Power, M. Foroozandeh, P. Moutzouri, R. W. Adams, M. Nilsson, S. R. Coombes, A. R. Phillips and G. A. Morris, Chem. Commun., 2016, 52, 6892 RSC; (b) Y. Cohen, L. Avram and L. Frish, Angew. Chem., Int. Ed., 2005, 44, 520 CrossRef CAS PubMed.
- (a) M. G. Sarwar, B. Dragisic, L. J. Salsberg, C. Gouliaras and M. S. Taylor, J. Am. Chem. Soc., 2010, 132, 1646 CrossRef CAS PubMed; (b) M. Erdélyi, Chem. Soc. Rev., 2012, 41, 3547 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods, analytical data, X-ray crystallographic details and additional data. CCDC 1909801–1909804. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc03061a |
| This journal is © The Royal Society of Chemistry 2019 |