
Tin-based nanomaterials: colloidal synthesis and battery applications
Xixia
Zhao
,
Qi
Yang
and
Zewei
Quan
 *
*
Department of Chemistry, Southern University of Science and Technology (SUSTech), Shenzhen, Guangdong 518055, P. R. China. E-mail: quanzw@sustech.edu.cn
First published on 12th June 2019
Abstract
Tin-based nanomaterials have been of increasing interest in many fields such as alkali-ion batteries, gas sensing, thermoelectric devices, and solar cells. Finely controllable structures and compositions of tin-based nanomaterials are crucial to improve their performances. The solution-based colloidal synthesis of these compounds offers a promising path toward controlling their structures and components. This feature article summarizes the progress in recent studies on the colloidal synthesis of tin-based nanomaterials (such as metallic tin, alloys, oxides, chalcogenides, and phosphides) and their applications in alkali-ion batteries including our own recent contributions to this subject. The challenges and future outlook of the controllable synthesis and practical development of tin-based anode materials are also addressed.
 Xixia Zhao | Xixia Zhao received his master's degree in chemical engineering and technology from China University of Petroleum (East China) in 2016 under the supervision of Prof. Changhua An. He continues to pursue a PhD over there under the supervision of Prof. Jun Zhang, and he has also been a visiting PhD student from 2016 in Prof. Zewei Quan's group at Southern University of Science and Technology. His current research topics focus on the controllable synthesis of inorganic nanomaterials for application in energy storage. |
 Qi Yang | Qi Yang is pursuing his BS degree in chemistry at Southern University of Science and Technology, where he has been conducting independent research projects under the supervision of Prof. Zewei Quan since 2016. His current research topics focus on the synthesis of colloidal tin-based alloy nanocrystals and their application in electrochemistry. |
 Zewei Quan | Zewei Quan is currently a professor in the Department of Chemistry at Southern University of Science and Technology in Shenzhen, China. He obtained his PhD in inorganic chemistry from Changchun Institute of Applied Chemistry, Chinese Academy of Sciences (with Prof. Jun Lin) in 2009. After that, he worked as a postdoctoral fellow and later as a research scientist at the State University of New York at Binghamton with Prof. Jiye Fang (2009–2012). He then joined Los Alamos National Laboratory as Oppenheimer Fellow (2012–2015). His current research interests include solution-phase synthesis, self-assembly, and high-pressure behavior of nanoparticles and their emerging applications. |
Introduction
Tin-based materials, including tin metal, alloys, oxides, chalcogenides, phosphides, and perovskites, are an important class of functional materials due to their earth-abundance, non-toxic nature, and intriguing physicochemical properties.1–5 As such, they have received widespread attention in the field of alkali-ion batteries, catalysis, gas sensors, solar cells, and solders.6–13 For example, tin possesses multiple valence states, good electrical conductivity, high mechanical and thermal stability, and the ability to form alloys with various alkali metals. Thus, tin and its various compounds are very promising host materials for the storage of different alkali-ions via the alloying/dealloying reactions between tin and alkali metals.6 As a promising anode in lithium-ion batteries, tin possesses a theoretical capacity of 994 mA h g−1, which is two times higher than that of the commercial graphite anode.14 In addition, tin dioxide is the most used semiconductor base material for toxic gas sensors, and tin-based selenides possess high chemical stability, narrow band gap (1.0–1.5 eV), and high absorption coefficient (∼105 cm−1) with p-type conductivity, which are essential for the efficient collection of solar radiation.7 Recently, tin-based perovskites have shown strong potential for use as absorbers in high-performance solar cells because of their electronic and optical characteristics which are comparable to those of well-developed lead-based perovskites.15,16Tin-based nanomaterials with well-designed structures have attracted researchers’ attention because of their enhanced performance for various applications. Compared with bulk materials, tin-based nanomaterials including metals and semiconductors usually exhibit distinctive properties.17–24 For example, the density of electronic states at Fermi surface in tin nanoparticles is increased compared with bulk tin, showing a larger heat capacity at low temperature.17 In addition, the melting point of metallic tin can be dramatically decreased when the particle size is reduced to nanoscale due to the high surface area to volume ratio for small tin particles.18,19 When the size of SnO2 nanocrystals approaches their exciton Bohr radius (2.7 nm), the charge carriers become spatially confined (electrons and holes move only in a potential well) and the coulomb shielding decreases. In this case, the exciton binding energy increases, the exciton effect becomes stronger, and more exciton absorption peak may appear, resulting in blueshift of the absorption edge relative to the bulk exciton absorption.20
To date, a variety of methods have been explored to prepare tin-based nanomaterials, including top-down and bottom-up approaches.25–28 The former aims to reduce the particle size by various mechanical forces, whereas the latter consists of the initial heterogeneous/homogeneous nucleation, followed by continuous growth. The top-down approach is versatile in production capability, but shows poor control of the structure/shape features of the final product. In contrast, bottom-up approaches, including using gas and liquid phase, exhibit good control of the nucleation/growth parameters.29,30 Among various bottom-up methods, colloidal synthesis has considerable advantages in the controlled synthesis of various tin-based nanoparticles by using long-chain organic compounds as capping ligands and/or solvents.5,31,32 The capping ligands play an important role in tuning their properties such as shape, crystalline structure, and electronic/optical properties, providing a possibility for the construction of complex structures. At the same time, as-prepared nanoparticles can also be engineered by ligand exchange to possess desirable properties, such as solubility and electrical conductivity. Colloidal synthesis has been proven to be a robust synthetic strategy for synthesizing tin-based nanomaterials with well-controlled size, shape, composition, and structure.
Applications of tin-based materials in energy storage can be dated back to the 1990s by Idota,33 who first reported the electrochemical performance of amorphous tin-based oxide as an anode for lithium-ion batteries. Subsequent studies indicate that tin can serve as a promising host for lithium storage through the formation of Li4.4Sn alloy with a much higher specific capacity than graphite.6 In addition, tin-based materials possess lower potential hysteresis than transition metal oxides, such as Fe2O3, Co3O4, and TiO2, which are also promising anode materials for lithium-ion batteries.34,35 Therefore, a variety of tin-based electrodes with well-designed structures have been developed to replace the low-capacity graphite anodes. Furthermore, since tin-based materials can electrochemically react with different alkali ions, they can also be used as versatile anode materials for some emerging battery systems, such as Na-, K-, and Mg-ion batteries.32,36–40
In this feature article, we summarize the recent advances in the colloidal synthesis of tin-based nanomaterials and their applications in alkali-ion (Li+, Na+, and K+) batteries, including our own recent contributions in this field. The first part is mainly focused on advances in the controlled synthesis of tin nanoparticles and various tin-based nanomaterials with specific compositions and delicate structures by the colloidal method. Then, the electrochemical properties of tin-based nanomaterials as anodes for different alkali-ion batteries are reviewed in the second part.
Colloidal synthesis of tin-based nanomaterials
Colloidal synthesis
In the past two decades, enormous developments of colloidal synthetic chemistry have made it possible to produce nanoparticles with well-controlled size, shape, composition and structure, offering researchers a new way to obtain nanomaterials with fascinating electrical, optical, and magnetic properties.41–51 Although the colloidal synthesis of nanoparticles can be performed in both aqueous and organic phase solutions, the organic phase synthesis is suitable to prepare many more types of high quality colloidal nanoparticles, especially those materials (including many tin-based nanoparticles) sensitive to O2 and H2O.52–54 During the organic phase synthesis of nanoparticles, organic reagents such as solvents and capping ligands are critical to control the size, shape and structure of nanoparticles. The synthesized colloidal nanoparticle usually presents an inorganic core with a monolayer of organic ligands.Considering the sensitivity of tin-based nanomaterials to O2/H2O during their preparation,27,32 organic phase synthesis is employed in our recent works to prepare various tin-based nanomaterials.5,12,13,55 Based on Fang's pioneering work on the synthesis of Pt and Pt-based binary alloys using tungsten hexacarbonyl (W(CO)6) as a reducing agent,56–58 we recently developed a generalized colloidal method to synthesize different types of uniform metal nanoparticles by reducing metal precursors in organic solution assisted by W(CO)6.59 On the basis of this method, we have synthesized uniform tin nanoparticles with tunable sizes. And then, a series of well-designed tin-based nanoparticles, including hollow tin oxide, hollow/yolk–shell tin chalcogenide, and binary alloy nanoparticles, have also been successfully synthesized via the template method. These developments will be discussed in detail in the following sections.
Colloidal synthesis of tin nanoparticles
Since tin has a low melting point (∼232 °C) and strong oxygen affinity, as well as due to its low affinity towards most commonly used surfactants, it is challenging to controllably synthesize high-quality tin nanoparticles, which always needs rigorous reaction conditions, including low temperature, strong reducing agents, and effective surfactants.32,60–66In the early 1990s, Bönnemann et al. first demonstrated the possibility of obtaining tin particles by reducing SnCl2 with LiBEt3H in tetrahydrofuran (THF) at room temperature.67 Later, Nayral et al. investigated the synthesis of tin nanoparticles (<100 nm) by the decomposition of (Sn(NMe2)2)2 in dry anisole.68 Then, Schlecht et al. tried to tune the size of tin nanoparticles (around 40–50 nm) by changing the concentration of the tin precursor (SnCl4).69 In contrast, Wang et al. reported the synthesis of tin nanoparticles with small sizes (3–19 nm) through the chemical reduction of a phenanthroline/tin chloride complex with NaBH4.70,71 Kwon et al. and Chee et al. further investigated the influence of capping agents and metallic precursors on the morphology and size of tin nanoparticles, respectively.72 Recently, Kravchyk et al. demonstrated the synthesis of tin nanoparticles with size control, in which monodisperse tin particles with different sizes varying from 9 to 23 nm were prepared using diisobutylaluminium hydride as a reducing agent in organic media.32
Our new method for synthesizing uniform tin nanoparticles was inspired by the recent success in using tungsten hexacarbonyl as a reducing agent for monodisperse metal nanoparticles.57 Monodisperse tin nanoparticles with three different sizes were readily synthesized by using tungsten hexacarbonyl as the reducing agent (Fig. 1).5 By changing the reaction temperature and time, uniform tin nanoparticles with average diameters of 11 nm, 13.5 nm, and 16 nm can be readily obtained. In addition, unlike traditional organometallic precursors used in previous reports, inexpensive and low-toxic SnCl2 was employed in this method, which is necessary to achieve large-scale preparation. The rapid decomposition of tungsten hexacarbonyl at elevated temperature (>140 °C) facilitates the quick nucleation of tin, and the presence of oleylamine plays a key role in both the crystallinity and uniformity of as-prepared tin nanoparticles.
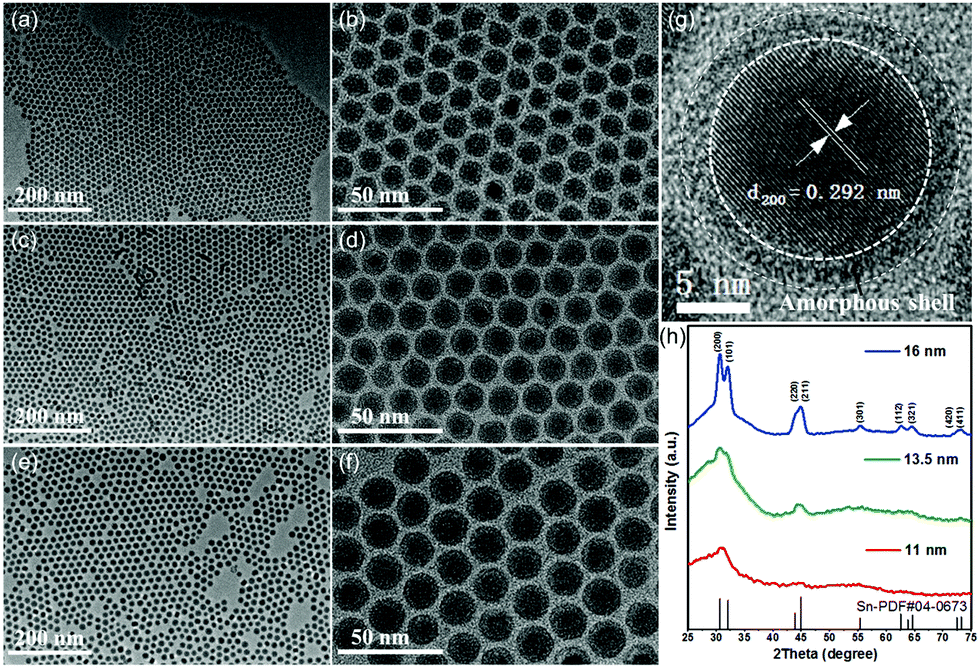 | ||
| Fig. 1 TEM images of 11 nm (a and b), 13.5 nm (c and d) and 16 nm Sn nanoparticles (e and f). (g) HRTEM image of an individual Sn nanoparticle and (h) XRD patterns of Sn nanoparticles with different diameters.5 | ||
Template synthesis of hollow tin-based oxide/chalcogenide nanoparticles
Tin-based compounds have broad application prospects in the fields of lithium-ion batteries, solar cells, and photocatalysis,1,73–75 some of which are highly dependent on their shapes. For example, the shape of nanocrystals with different exposed surfaces determines the electrical response and selectivity of SnO2 chemical sensors.76 Compared to their bulk counterparts, tin-based nanomaterials with well-defined shapes, such as 1D SnO2 nanowires77 and 2D SnO nanosheets,78 possess diminutive size and high surface-to-volume ratio, which can therefore effectively alleviate the detrimental effect of volume change and shorten the diffusion length of lithium ions upon lithiation/delithiation. Due to these attractive properties of tin-based nanomaterials, the delicate preparation of tin-based nanomaterials has become one of the hot topics in materials research. Among different architectures, hollow nanoparticles with controllable hollow interior and shell thickness are an important kind of nanostructured materials because of their large surface area and low material density. Since the first synthesis of CoS and CoO hollow nanoparticles through the sulfidation and oxidation of Co nanoparticles in 2004,79 several different types of hollow nanoparticles have been prepared by similar methods based on the nanoscale Kirkendall effect.80The imbalance of diffusion rates between two components is a feature of the Kirkendall effect. Typically, a diffusion couple would be generated when metal nanoparticles are exposed to precursors such as sulfur, selenium, oxygen, and phosphorus at high temperatures. If the outward diffusion of metal cations is much faster than the inward diffusion of anions, the vacancies can be formed and flow inward along with the outward flux of metal cations to balance this difference. When these vacancies become oversaturated, they would coalesce into voids and finally result in the formation of hollow nanoparticles containing two elements.81 In addition, the Ostwald ripening process is also a common strategy to fabricate hollow structures, which involves the growth of larger crystals at the expense of smaller ones.82
Our group first reported the in situ conversion of uniform Sn nanoparticles to hollow tin oxide nanospheres and yolk–shell/hollow tin chalcogenides (SnS, SnSe, and SnTe) based on the nanoscale Kirkendall effect (Fig. 2).5 We proposed reasonable formation mechanisms of these hollow structures. As shown in Fig. 2b, in the initial stage of in situ oxidation, the surface of the Sn nanoparticle first reacts with oxygen to produce a thin tin-oxide shell, and further oxidation reaction occurs through the diffusion of atoms at the interface. Since the outward diffusion of Sn atoms (JSn) at the interface is faster than the inward diffusion of O atoms (JO), small vacancies are formed at the interface. Subsequently, these small vacancies further coalesce to form hollow SnOx nanospheres.
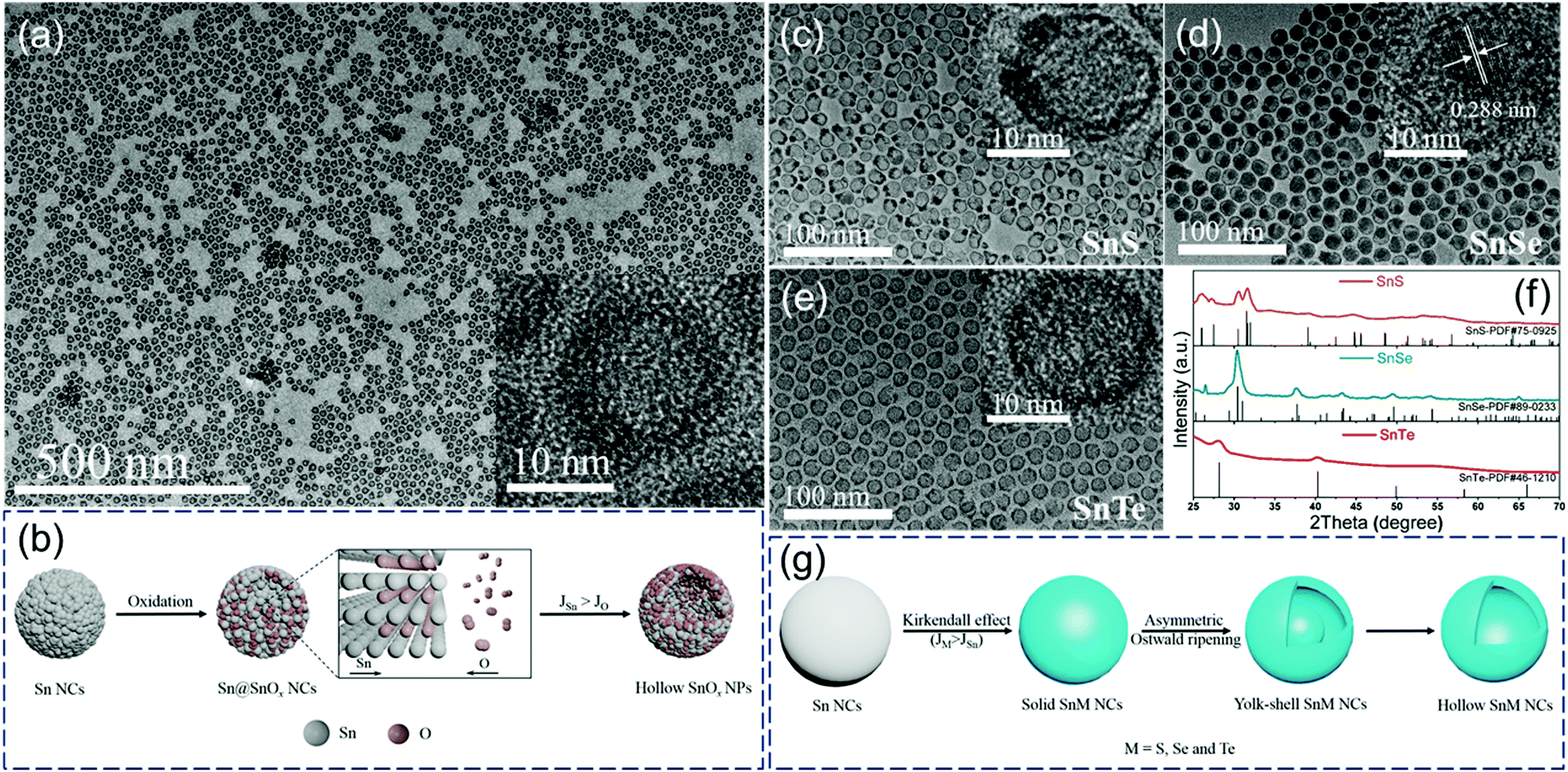 | ||
| Fig. 2 (a) TEM images of hollow SnOx nanospheres and (b) schematic illustration of the synthesis of hollow SnOx nanospheres. TEM images (c–e) and XRD patterns (f) of hollow SnS, yolk–shell SnSe and hollow SnTe nanoparticles. (g) Scheme of the synthesis of hollow SnS, yolk–shell SnSe and hollow SnTe nanoparticles.5 | ||
In contrast, the formation process of tin-based chalcogenide (SnS, SnSe, and SnTe) nanoparticles is slightly different from the oxidation process. As shown in Fig. 2g, a two-step conversion process including the Kirkendall effect and asymmetrical Ostwald ripening process occurs when Sn reacts with S, Se, and Te. Solid tin-based chalcogenide nanoparticles are first formed because of the faster inward diffusion of chalcogen atoms compared with the outward diffusion of Sn atoms at the interface. With the extension of reaction time, an asymmetric Ostwald ripening process, including core dissolution and shell recrystallization, takes place in regions consisting of smaller or looser grains, resulting in the formation of yolk–shell/hollow structures (Fig. 2c–e).
Template synthesis of tin-based binary alloy nanoparticles
Compared with single metals, alloys consisting of two or more metals usually have improved properties, such as higher hardness, lower melting point, and corrosion resistance.83 Since tin can alloy with many metals to form different tin-based alloys with distinctive properties,84 the synthesis of tin-based alloy nanoparticles with tunable composition is intensively explored. It has been proved that the catalytic, electrical, and optical properties of some metal nanoparticles can be significantly changed by combining them with tin, as in the case of SnGe and PtSn alloys.85–87 In addition, the fabrication of tin-based alloy anodes (e.g., SnSb, SnCo) for lithium-ion batteries can effectively alleviate their volume expansion during charge and discharge processes.88–90Due to the narrow bandgap and good near-infrared-emitting property, SnGe alloys are a kind of ideal candidates for infrared optoelectronics and optical devices, such as infrared lasers, photodetectors, or light-emitting diodes.91–95 In addition, the addition of Sn to the Ge matrix also leads to enhanced electron and hole mobility, making SnGe alloys attractive for high-speed electronic devices.96 However, the synthesis of uniform SnGe alloy nanoparticles is quite difficult due to the high crystallization temperature of Ge, their low solubility (<1%), as well as a large lattice mismatch (∼14%) between Sn and Ge.97
Our group recently demonstrated a novel colloidal method to prepare uniform Sn1−xGex alloy nanoparticles through the reaction between the Sn nanoparticle template and the Ge precursor under reducing conditions (Fig. 3).61 By fine-tuning the reaction temperature and time, uniform Sn1−xGex alloy nanoparticles with a tunable bandgap from 0.51 to 0.72 eV were successfully synthesized. Notably, compared with previously reported methods (above 300 °C), this method can occur at a much lower temperature (60–180 °C) ascribed to the catalytic effect of Sn seeds on reducing the reaction energy barriers.98 Through ex situ characterization of samples collected at different reaction stages, an evolution mechanism from Sn nanoparticles to Sn1−xGex alloy nanoparticles is proposed based on the galvanic replacement and the solid diffusion mechanism, as shown in Fig. 3j. Sn nanoparticles first react with the Ge precursor under a reducing condition to form an amorphous Sn1−xGex shell on the surface. Then, the amorphous Sn1−xGex shell gradually transforms to the cubic phase Sn1−xGex alloy with increasing temperature. Further solid diffusion process results in the final formation of homogeneous Sn1−xGex alloy nanoparticles.
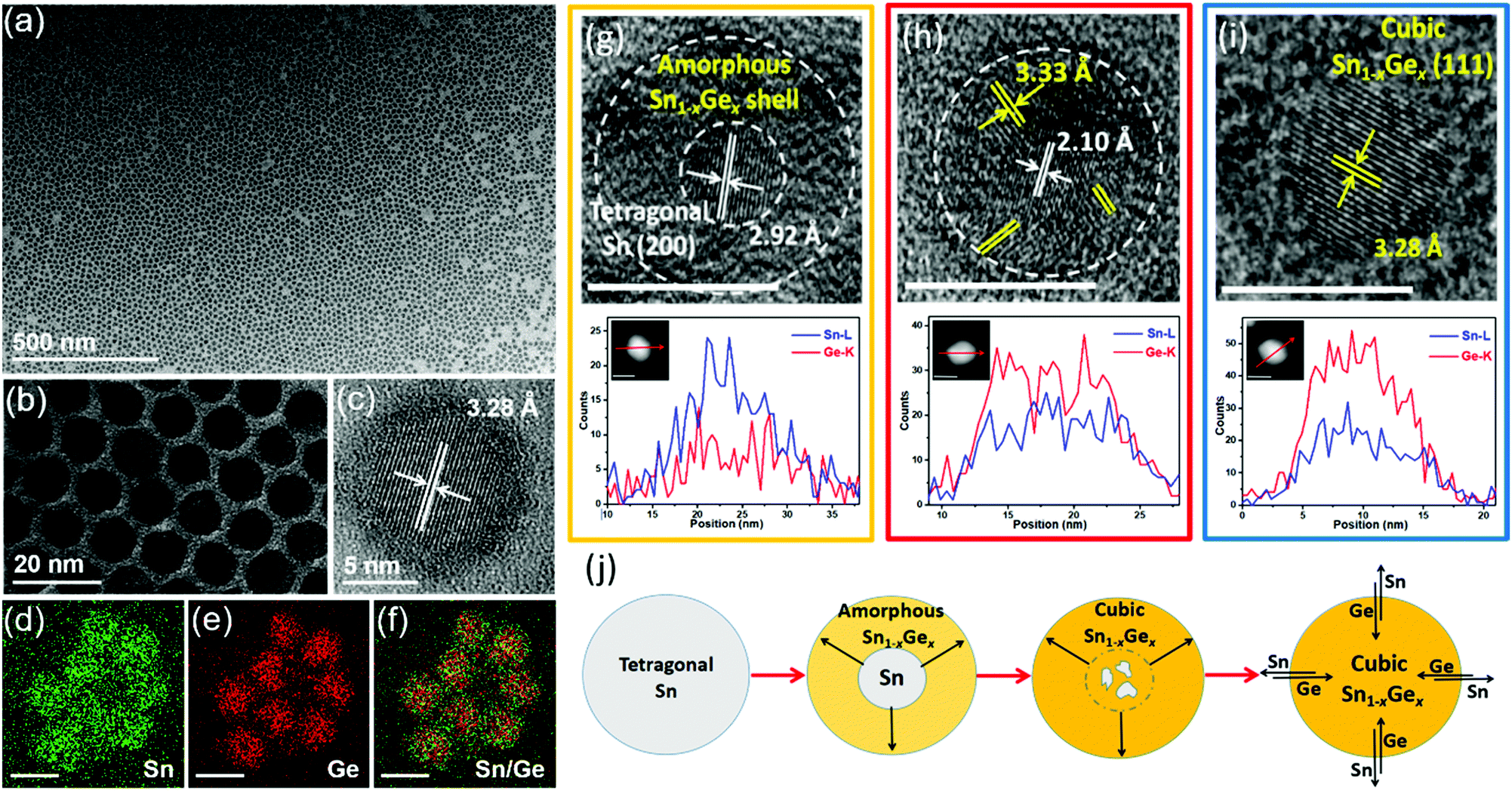 | ||
| Fig. 3 (a–c) TEM and HRTEM images of as-prepared Sn0.18Ge0.82 alloy nanoparticles prepared at 180 °C for 5 h. (d–f) EDS elemental mapping results of Sn0.18Ge0.82 nanoparticles. The scale bars in (d–f) represent 10 nm. HRTEM images, STEM images, and the corresponding EDS elemental line scan profiles of samples collected at different reaction stages: (g) Sn-amorphous Sn1−xGex heterostructure collected at 90 °C, (h) Sn-crystalline Sn1−xGex heterostructure collected at 120 °C, and (i) Sn0.18Ge0.82 alloy nanoparticles prepared at 180 °C for 5 h. The scale bars represent 10 nm. (j) Schematic illustration of the transformation process from Sn nanoparticles to Sn1−xGex alloy nanoparticles. Reprinted with permission from ref. 61. Copyright 2019 American Chemical Society. | ||
This robust method is being extended to prepare other novel tin-based alloy materials by our group, such as Sn1−xSbx and Sn1−xBix alloys. Similarly, Jiao's group also reported the synthesis of AgSn alloy nanoparticles, including the formation of Sn seeds and a galvanic replacement between Sn and Ag.7 The composition of AgSn alloys can be tuned accordingly by changing the molar ratio of Sn and Ag precursors.
Applications of tin-based nanomaterials in batteries
Tin-based nanomaterials have stimulated a growing interest in many fields owing to their useful physicochemical properties. For example, SnO2 is investigated as a gas sensor material due to its high gas sensitivity.99 Moreover, SnSe is considered as a novel type of promising thermoelectric material with ultralow thermal conductivity and high thermoelectric figure of merit.100 In addition, the tin-based perovskite is an alternative low-toxic photovoltaic material with an ideal band gap (∼1.3 V) compared with the lead-based perovskite.2 Moreover, tin-based nanomaterials possess huge potential as versatile high-performance anodes for alkali-ion (Li+, Na+, K+) batteries, because tin can act as a host material for the storage of all these alkali-ions via electrochemical alloying/de-alloying with them.6As is known, large volume change of tin-based anode materials during the charge/discharge process would result in their pulverization and delamination from current collectors, leading to rapid capacity fading and poor rate performance. It has been proved that the size, structure, phase, and composition of tin-based nanomaterials play a great role in improving their performances.1,6 For example, reducing the particle size and introducing hollow/porous structures can effectively accommodate the volume changes of tin-based anode materials by relieving mechanical stress and providing extra space.32 Moreover, fabrication of amorphous tin-based structures or mixing tin with other metals to form alloys is also an effective way to achieve improved electrochemical performances.88–90
In this section, we focus on several types of tin-based nanomaterials (e.g., metallic tin, oxides, selenides, and phosphides) that are delicately designed for alkali-ion battery anodes including our recent works, and discuss the influence of size, structure, and composition on their electrochemical performance.
Lithium-ion battery
Tin-based materials can alloy with lithium and deliver higher capacities than graphite. However, their huge volume change during charging and discharging can lead to pulverization of the electrode and result in the failure of the whole battery. Previous studies have shown that reducing the size of tin-based materials is beneficial to adapt the mechanical stress caused by the alloy/de-alloy process, thus significantly improving their electrochemical properties. For example, Kravchyk et al.32 reported that tin nanoparticles of ∼10 nm can show a reversible capacity of 300 mA h g−1 for 100 cycles at a current density of 1000 mA g−1, which is significantly better than that of commercial Sn nanoparticles (50–150 nm). Furthermore, Cabana et al. studied the effect of particle size on the volume change of tin during lithiation using ex situ transmission electron microscopy (TEM).101 They found that reducing the particle size can effectively alleviate the cracking of tin nanoparticles, although slight particle pulverization still occurs.Recently, our group demonstrated the key role of small size and an oxide layer in improving the cycle stability of tin nanoparticles (Fig. 4).12 As-prepared Sn nanoparticle electrodes delivered high reversible capacities during long cycling. TEM images after cycling (Fig. 4c and d) revealed that the morphology of Sn nanoparticles after cycling is the same as that of the pristine one, indicating that small size plays a crucial role in alleviating the detrimental effect of volume expansion during repeated charge/discharge processes, as the mechanical stress induced by the volume change during the alloying/dealloying process can be accommodated more easily in these tiny nanoparticles,32,101 consistent with their good cycle stability. Meanwhile, in the initial irreversible discharge reaction, the oxide layer on tin nanoparticles reacted with lithium to form a Li2O matrix, which is conducive to maintaining the structural stability of the electrode material and further improving its cycle performance.
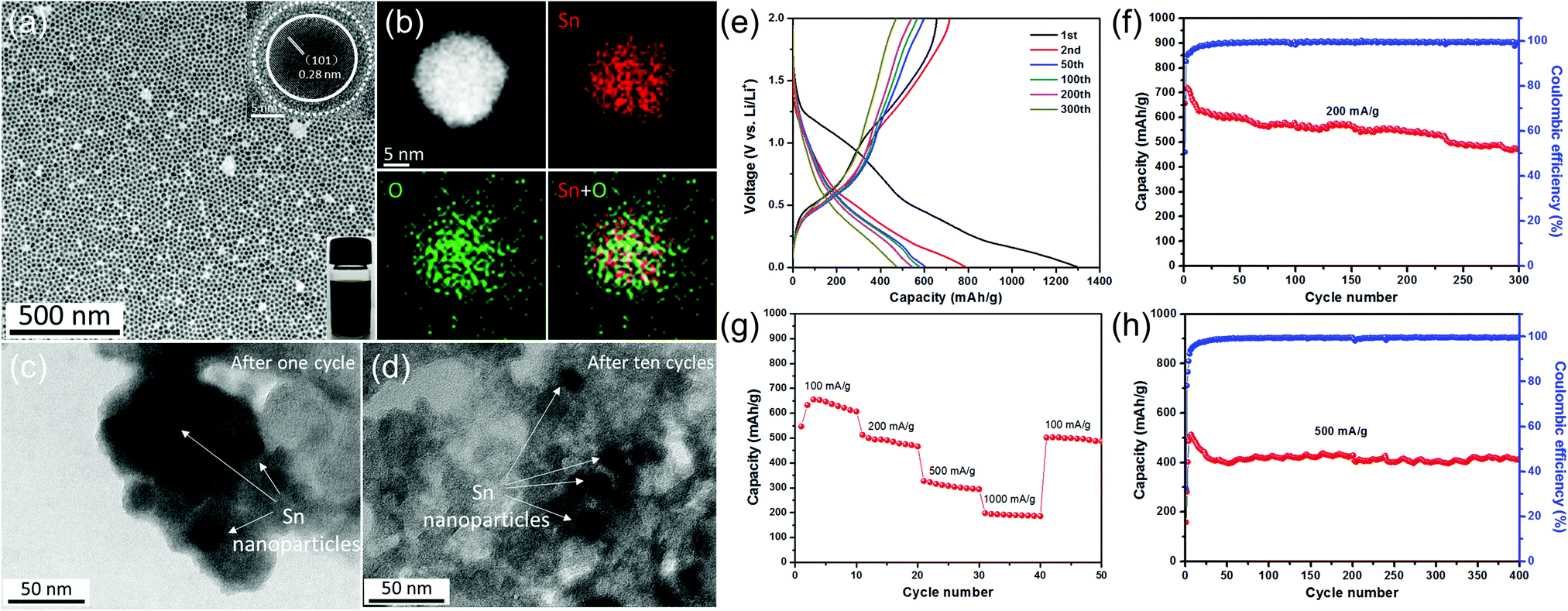 | ||
| Fig. 4 (a) TEM image of the as-prepared Sn nanoparticles and (b) the STEM image and EDX elemental mapping of a single Sn nanoparticle. TEM images of as-prepared Sn nanoparticles (c) after one cycle and (d) after ten cycles at a current density of 500 mA g−1. (e) Selected galvanostatic charge/discharge voltage profiles at 200 mA g−1. Cycling performance and the corresponding coulombic efficiency (f) at 200 mA g−1 and (h) at 500 mA g−1. (g) Rate capability from 100 to 1000 mA g−1.12 | ||
Apart from metallic tin, tin oxides have also been considered as a kind of promising anode candidates for high-performance lithium-ion batteries due to their considerable theoretical capacities (SnO2, 782 mA h g−1). However, similar to the tin anode, the large volume change during lithiation/de-lithiation cycles is also a significant problem of the tin oxide anode. Introduction of a hollow or porous structure into tin oxide anode materials is an effective way to improve their electrochemical properties. This is because the hollow or porous structure can not only accommodate large volume changes, but also shorten the diffusion path of lithium ions.102–104 In addition, the effect of the crystallinity of SnO2 on the lithium-storage performance has also been studied. The results show that compared with the crystalline structure, amorphous SnO2 is widely believed to more effectively overcome the huge volume change of the electrode due to its isotropic volume expansion during the charge/discharge processes, thus delaying the capacity fading.105,106
Based on the above considerations, we expect that the rational design of tin oxide nanoparticles with a hollow/porous structure and amorphous properties tends to achieve improved electrochemical performance. Therefore, our group elaborately designed and synthesized a kind of amorphous SnOx hollow nanospheres by a template method based on the nanoscale Kirkendall effect (Fig. 5).12 As expected, the amorphous SnOx hollow nanospheres exhibited high reversible capacities and excellent rate properties as an anode for lithium-ion batteries, which can be attributed to the small size, hollow/amorphous structure, and surface formation of the Li2O matrix. These structural features not only accommodate the huge volume change of the material during the charge/discharge processes, but can also help to shorten the diffusion distance of lithium ions.
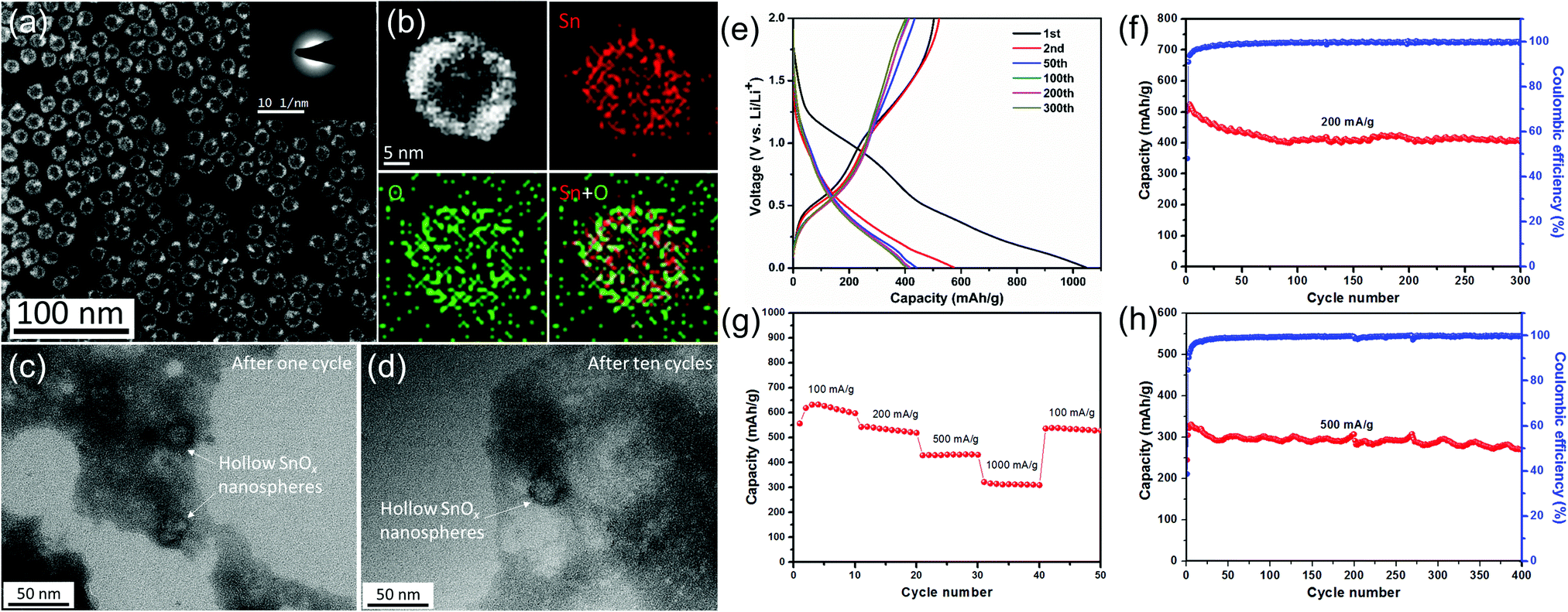 | ||
| Fig. 5 (a) STEM image of the as-prepared hollow SnOx nanospheres and (b) the STEM image and EDX elemental mapping of a single hollow SnOx nanosphere. TEM images of as-prepared hollow SnOx nanospheres (c) after one cycle and (d) after ten cycles at a current density of 500 mA g−1. (e) Selected galvanostatic charge/discharge voltage profiles at 200 mA g−1. Cycling performance and the corresponding coulombic efficiency (f) at 200 mA g−1 and (h) at 500 mA g−1. (g) Rate capability from 100 to 1000 mA g−1.12 | ||
Sodium-ion battery
As a promising alternative to lithium-ion batteries, sodium-ion batteries are considered to have broad application prospects in the field of large-scale energy storage, mainly due to their low-cost, abundant reserves and even distribution of sodium resources.107–109 However, their practical application is greatly limited due to lack of reliable anodes, and thus it is urgent to develop sodium-ion battery anode materials with high capacities and long cycle life. So far, tin, antimony, phosphorus and other materials have been investigated as the anode of sodium-ion batteries, and have exhibited potential in electrochemical sodium storage.110,111Due to its high theoretical capacity (∼780 mA h g−1), good chemical stability, and abundant resources, tin selenide (SnSe) has become a potential anode material for sodium-ion batteries in recent years.112,113 However, similar to other tin-based materials, SnSe as anode material for sodium-ion batteries also has similar problems such as the volume change and agglomeration of SnSe particles, further leading to rapid capacity fading and poor rate performance. To solve these problems, the regulation of the shape/size of SnSe particles to mitigate volume expansion and agglomeration or the addition of conductive substrates (such as carbon fiber and graphene) into SnSe particles has been widely adopted, achieving significantly improved electrochemical sodium storage performance.114,115
To improve the electrochemical sodium storage performance of SnSe, we first designed and synthesized monodisperse SnSe nanoparticles with a unique yolk–shell structure by a template method (Fig. 6).13 The unique yolk–shell structure and ultra-small particle size can not only shorten the diffusion distance of sodium ions, but can also alleviate the volume change and the aggregation of SnSe nanoparticles in the charge and discharge processes, thus leading to enhanced electrochemical performance. When such SnSe nanoparticles were used as the anode for sodium-ion batteries, they can deliver an initial capacity of 355.8 mA h g−1 and a capacity retention rate of 72.5% after 150 cycles at 500 mA g−1. In addition, they also showed excellent rate performance: the capacity retention rate is as high as 85.3% at a current density of 1000 mA g−1, compared with the capacity at 100 mA g−1. Quantitative analysis demonstrated that the capacitive contribution dominates the total sodium storage performance of our SnSe electrodes, which also explains their superior rate performance.
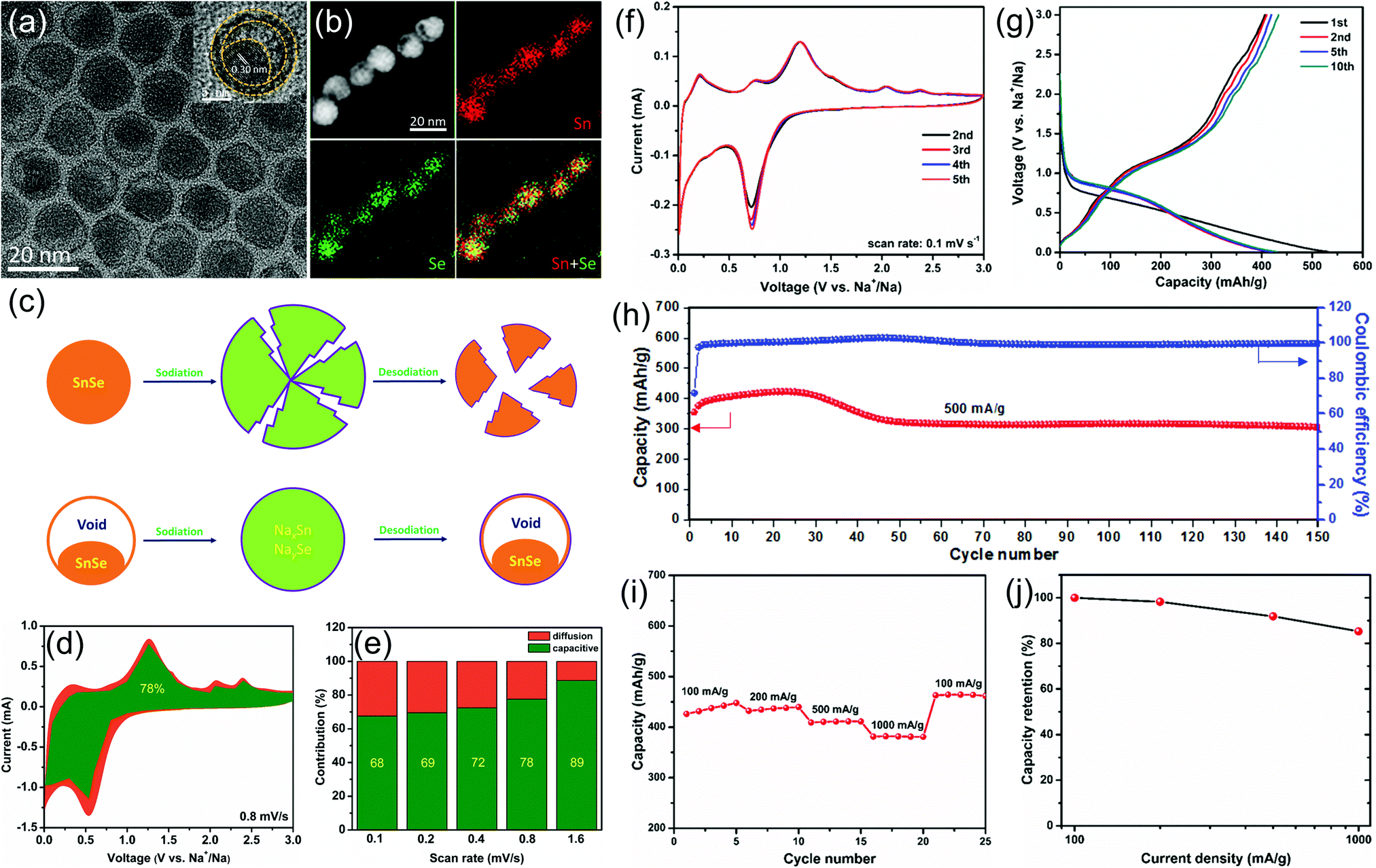 | ||
| Fig. 6 (a) TEM images, (b) STEM image and the corresponding EDX elemental mapping of Sn and Se and their overlapped image. The inset of (a) shows the high resolution TEM image of a single SnSe nanoparticle. (c) Schematic illustration to show the advantages of yolk–shell structured SnSe nanoparticles during the sodiation/desodiation process. (d) Capacitive (green) and diffusion-controlled (yellow) contributions to charge storage of the as-prepared SnSe nanoparticles at 0.8 mV s−1. (e) Normalized contribution ratio of capacitive (green) and diffusion-controlled (yellow) capacities at different scan rates. (f) Typical CV curves recorded at a scan rate of 0.1 mV s−1. (g) Selected galvanostatic charge/discharge voltage profiles at 200 mA g−1. (h) Cycling performance and the corresponding coulombic efficiency at 500 mA g−1. (i) Rate capability and (j) capacity retention from 100 to 1000 mA g−1.13 | ||
Potassium-ion battery
Due to the abundance and low-cost of potassium resources compared to lithium resources, potassium-ion batteries as a possible energy system have gradually attracted researchers' attention, among which the potassium ion is considered as the working medium to drive this new “rocking chair” battery.116,117 In addition, due to the lower standard redox potential of potassium in non-aqueous electrolytes compared to that of sodium, potassium-ion batteries are more likely to construct low-cost, high-energy density and high-voltage storage systems.118Although graphite is considered as a promising anode material for potassium-ion batteries, construction of high-energy density potassium-ion batteries based on the graphite anode is challenging because of its relatively low capacity. Therefore, it is necessary to explore other anode materials with higher capacities.11 Tin phosphide materials are a member of burgeoning anode candidates for potassium-ion batteries due to their high theoretical capacities and much higher electronic conductivity compared to pure phosphorus.119,120 Meanwhile, a synergistic potassium storage mechanism may occur in tin phosphide anodes, where Sn and P elements can react with K to form K–Sn and K–P alloys, respectively. The resulting K–Sn alloy can act as a conducting pathway to activate the reversible K-storage/release of nonconductive K–P phases, and the well-dispersed K–P phase would serve as a matrix to prevent the agglomeration of K–Sn/Sn.121 These characteristics together render tin phosphide to be a promising anode for potassium-ion batteries. Zhang et al.122 reported that Sn4P3–C composite material can be used as a high-capacity anode material for potassium-ion batteries, showing obvious advantages over pure phosphorus. Later, they further proposed a synergistic K-storage mechanism of Sn4P3 including a sequential conversion from P to K3P11/K3P and the alloying reaction between Sn and K to form KSn.119 However, current investigations are limited to Sn4P3, and there are few reports about phosphorus-richer tin phosphides as anodes for potassium-ion batteries, which possess much higher theoretical capacities.
Our group recently developed a simple colloidal method to prepare the phosphorus-richer SnP0.94 nanoplates. To further improve their stability, the SnP0.94@GO composite was fabricated by combining SnP0.94 nanoplates with graphene oxide (GO) sheets (Fig. 7).123 As a new type of anode material for potassium-ion batteries, SnP0.94@GO composite material prepared by us has shown good electrochemical properties. At a current density of 25 mA g−1, the SnP0.94@GO electrode showed an initial depotassiation capacity of 294 mA h g−1, and a high-capacity retention rate of 83% after 50 cycles. Even at 200 mA g−1, the composite could deliver a depotassiation capacity of 106 mA h g−1, and maintain 93% of capacity after 100 cycles. The good potassium storage performance of the SnP0.94@GO electrode is attributed to the synergetic effects of the high theoretical capacity of SnP0.94 and GO sheet matrix that could buffer volume change, accelerate charge transfer, and reduce contact between active materials and electrolytes.
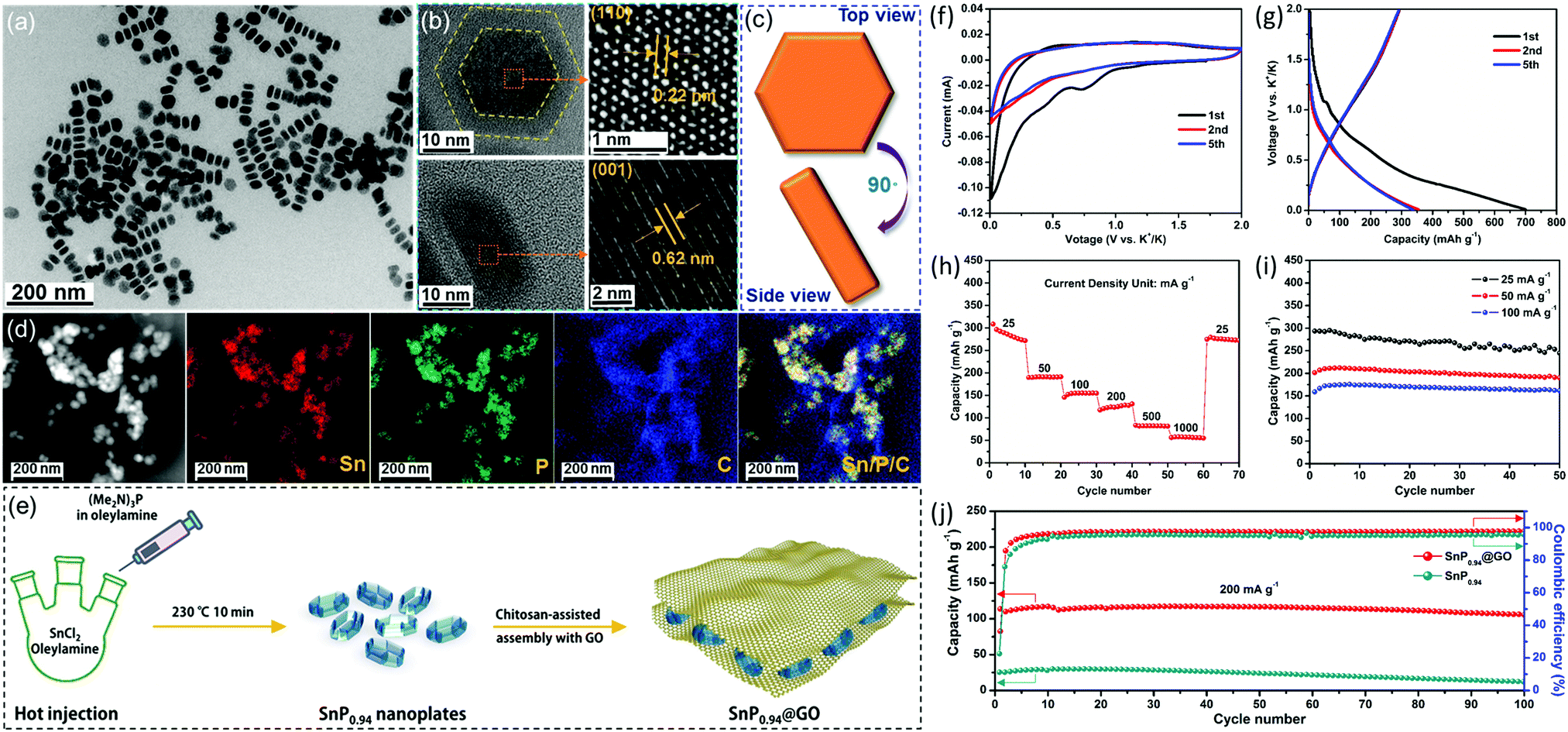 | ||
| Fig. 7 (a) TEM image of as-synthesized SnP0.94 nanoplates. (b) HRTEM images and (c) schematic diagram of a typical SnP0.94 nanoplate viewed from top and side. (d) STEM image and EDS mapping images of the as-prepared SnP0.94@GO composite. (e) Schematic depicting the formation of the SnP0.94@GO composite. (f) Typical CV curves recorded with a scan rate of 0.1 mV s−1. (g) Selected galvanostatic charge/discharge voltage profiles at 25 mA g−1. (h) Rate capability from 25 to 1000 mA g−1. (i) Cycle performances at different current densities. (j) Cycle performances and the corresponding coulombic efficiencies of the SnP0.94 nanoplates and SnP0.94@GO composite at a current density of 200 mA g−1. Reprinted with permission from ref. 123. Copyright 2019 Elsevier. | ||
Table 1 summarizes the recent advances in the performance of some typical tin-based nanomaterials as anodes for Li-, Na-, and K-ion batteries including our own recent contributions to this subject. Compared with traditional graphite anode materials, tin-based anode materials have some advantages such as higher mass specific capacity and higher lithiation/delithiation potential. At the same time, these tin-based anode materials usually suffer from poor cycling stability due to their larger volume changes. This stability issue can be improved by delicately designing their size/structure as discussed herein or combining them with other highly stable materials, such as carbon and graphene. It is expected that well-designed tin-based nanomaterials can deliver superior performance as anodes for different types of alkali-ion batteries.
| Battery type | Material | Initial cycle performance | Cycling performances | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| Initial charge capacity (mA h g−1) | Initial coulombic efficiency (%) | Current density (mA g−1) | Capacity retention (mA h g−1) | Number of cycles | Current density (mA g−1) | |||
| Li-Ion battery | 3D porous graphene networks anchored with Sn nanoparticles | 1245 | 69 | 200 | 682 | 1000 | 2000 | 124 |
| Nano-Sn/C composite | 710 | 69 | 200 | 710 | 130 | 200 | 125 | |
| Yolk–shell SnO2@C nanoparticles | 1236 | 56.4 | 100 | 630 | 100 | 100 | 126 | |
| Coaxial SnO2@C hollow nanospheres | 730 | 49.6 | 200 | 460 | 100 | 500 | 127 | |
| SnSe/C nano composite | 412.4 | 55.1 | 500 | 324.6 | 200 | 500 | 114 | |
| Monodisperse Sn nanoparticles | 657.2 | 51 | 200 | 416.4 | 400 | 500 | 12 | |
| Hollow/amorphous tin oxide nanospheres | 503.0 | 48 | 200 | 402.5 | 300 | 200 | 12 | |
| Na-Ion battery | 1–3 nm thick SnO on carbon cloth | 848 | 79.1 | 100 | 452 | 1000 | 1000 | 128 |
| Carbon covered SnS/SnO2 heterostructure anchored to graphene | 729 | 74.6 | 30 | 409 | 500 | 810 | 129 | |
| SnS/3D N-doped graphene hybrid | 890.5 | 80.1 | 100 | 509.9 | 1000 | 2000 | 130 | |
| SnS2 nanocrystals on amino-functionalized graphene | 749 | 73 | 200 | 480 | 1000 | 1000 | 131 | |
| Single-crystalline SnSe nanosheet clusters | 738 | ∼95 | 25 | 271 | 100 | 200 | 112 | |
| Ultrathin layered SnSe nanoplates | 738 | 79.3 | 50 | 312.7 | 300 | 2000 | 113 | |
| Yolk–shell structured SnSe nanoparticles | 406.0 | 76.5 | 200 | 307.1 | 150 | 500 | 12 | |
| K-Ion battery | 3D carbon/Sn | 384.5 | 45.3 | 50 | 276.4 | 100 | 50 | 132 |
| SnSb–graphene–carbon nanofibers | 320.6 | 52.5 | 100 | 275.1 | 100 | 100 | 133 | |
| Carbon confined SnO2 | 195 | 28 | 50 | 122 | 200 | 100 | 134 | |
| SnO2–graphene–carbon nanofibers | 229.2 | 44.1 | 100 | 202 | 100 | 100 | 135 | |
| SnS2–rGO | 355 | 56.3 | 25 | 250 | 30 | 25 | 136 | |
| Sn4P3/C | ∼350 | 59.5 | 50 | 307.2 | 50 | 50 | 122 | |
| Sn4P3@carbon fiber | ∼510 | 64.1 | 50 | 160.7 | 1000 | 500 | 119 | |
| SnP0.94 nanoplates/graphene oxide composite | 294 | 42 | 25 | 162 | 50 | 100 | 123 | |
Conclusions and outlook
Tin-based nanomaterials have great application prospects in many fields, and their performance is greatly defined by their structure and composition. Colloidal synthesis is a powerful synthetic strategy and has been successfully applied for controllably synthesizing tin-based nanomaterials. In this feature article, we have focused on the developments from our group in colloidal synthesis and application in batteries of tin-based materials. We have first summarized recent advances in the colloidal synthesis of tin nanoparticles and other tin-based nanomaterials such as oxides, chalcogenides, and alloys. These materials exhibit delicate and well-designed structures and compositions, which play crucial roles in determining their applications. We have also discussed the electrochemical performance of several types of tin-based nanomaterials, including metallic tin, oxides, selenides, and phosphides, in different alkali-ion (Li+, Na+, and K+) batteries.Despite these exciting developments, some critical issues still need to be addressed for the further development of tin-based nanomaterials. In our opinion, several main concerns are listed as below:
(1) Controlled synthesis provides an effective strategy to optimize size- and shape-dependent properties of metals. Nevertheless, rigorous shape control of tin nanoparticles in colloidal synthesis still remains a great challenge due to the lack of ligands that can selectively adsorb on specific crystal surfaces of tin particles to control their growth in certain directions. To further advance this field, looking for appropriate ligands to facilitate the anisotropic growth of tin nanoparticles would be greatly desired.
(2) Organic capping ligands play a crucial role in the size- and shape-control of tin-based nanomaterials and would finally absorb on the particle surface, enabling their excellent solubility in nonpolar solvents. However, these organic capping ligands usually possess poor electrical conductivity due to their long organic chains, greatly affecting the electrochemical performance of tin-based nanomaterials. Therefore, removing them or replacing them with shorter ones is very necessary to improve their electrical conductivity. However, these post-treatment processes inevitably complicate the preparation of tin-based nanomaterials. It would be also meaningful if these ligands on the particle surface can be in situ carbonized into a conductive carbon shell or matrix with the promise that the as-prepared tin-based nanomaterials are not destroyed.
(3) To date, the cycle performance and rate capacity of tin-based materials have been significantly improved by controlling their size, structure, and composition. However, some key electrochemical parameters are still not up to commercial standards. For example, due to the large specific surface area, tin-based nanomaterials usually consume plenty of alkali ions in the formation process of a thick SEI layer, resulting in a large loss of initial capacity. Therefore, more work needs to be done to exploit modification methods for achieving higher initial coulombic efficiency. In addition, the larger radii of sodium and potassium would cause larger volume changes and slower kinetics of tin-based anode materials during the charge/discharge processes, leading to great challenges in the structural design and morphological control of tin-based anode materials.
In summary, the electrochemical performance of tin-based materials can be significantly improved by regulating their size, structure, and composition during colloidal synthesis. Meanwhile, more efforts are required to design tin-based materials with targeted properties that could overcome current limitations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (No. 51772142), the Development and Reform Commission of Shenzhen Municipality (Novel Nanomaterial Discipline Construction Plan), and the Shenzhen Science and Technology Innovation Committee (JCYJ20170412152528921, KQJSCX20170328155428476, KQTD2016053019134356).Notes and references
- L. Liu, F. Xie, J. Lyu, T. Zhao, T. Li and B. G. Choi, J. Power Sources, 2016, 321, 11–35 CrossRef CAS.
- W. Ke, C. C. Stoumpos and M. G. Kanatzidis, Adv. Mater., 2018, 1803230 CrossRef PubMed.
- Q. Li, J. Fu, W. Zhu, Z. Chen, B. Shen, L. Wu, Z. Xi, T. Wang, G. Lu, J. J. Zhu and S. Sun, J. Am. Chem. Soc., 2017, 139, 4290–4293 CrossRef CAS PubMed.
- N. H. Chou and R. E. Schaak, Chem. Mater., 2008, 20, 2081–2085 CrossRef CAS.
- X. Zhao, Q. Di, X. Wu, Y. Liu, Y. Yu, G. Wei, J. Zhang and Z. Quan, Chem. Commun., 2017, 53, 11001–11004 RSC.
- B. Huang, Z. Pan, X. Su and L. An, J. Power Sources, 2018, 395, 41–59 CrossRef CAS.
- N. Koteeswara Reddy, M. Devika and E. S. R. Gopal, Crit. Rev. Solid State Mater. Sci., 2015, 40, 359–398 CrossRef CAS.
- Z. Li, J. Ding and D. Mitlin, Acc. Chem. Res., 2015, 48, 1657–1665 CrossRef CAS PubMed.
- C. C. Stoumpos, C. D. Malliakas and M. G. Kanatzidis, Inorg. Chem., 2013, 52, 9019–9038 CrossRef CAS PubMed.
- W. J. Boettinger, C. E. Johnson, L. A. Bendersky, K. W. Moon, M. E. Williams and G. R. Stafford, Acta Mater., 2005, 53, 5033–5050 CrossRef CAS.
- W. Luc, C. Collins, S. Wang, H. Xin, K. He, Y. Kang and F. Jiao, J. Am. Chem. Soc., 2017, 139, 1885–1893 CrossRef CAS PubMed.
- X. Zhao, W. Wang, Z. Hou, Y. Yu, Q. Di, X. Wu, G. Wei, Z. Quan and J. Zhang, Inorg. Chem. Front., 2019, 6, 473–476 RSC.
- X. Zhao, W. Wang, Z. Hou, X. Fan, G. Wei, Y. Yu, Q. Di, Y. Liu, Z. Quan and J. Zhang, Inorg. Chem. Front., 2019, 6, 562–565 RSC.
- Y. Kim, K. H. Ha, S. M. Oh and K. T. Lee, Chem. – Eur. J., 2014, 20, 11980–11992 CrossRef CAS PubMed.
- L. Mao, H. Tsai, W. Nie, L. Ma, J. Im, C. C. Stoumpos, C. D. Malliakas, F. Hao, M. R. Wasielewski, A. D. Mohite and M. G. Kanatzidis, Chem. Mater., 2016, 28, 7781–7792 CrossRef CAS.
- B. Lee, C. C. Stoumpos, N. Zhou, F. Hao, C. Malliakas, C. Y. Yeh, T. J. Marks, M. G. Kanatzidis and R. P. Chang, J. Am. Chem. Soc., 2014, 136, 15379–15385 CrossRef CAS PubMed.
- A. A. Shikov, G. K. Panova, M. G. Zemlyanov, P. P. Parshin, Y. A. Kumzerov, A. A. Naberezhnov and D. S. Shaitura, Phys. Solid State, 2011, 53, 2515–2519 CrossRef CAS.
- H. Jiang, K. S. Moon, H. Dong, F. Hua and C. P. Wong, Chem. Phys. Lett., 2006, 429, 492–496 CrossRef CAS.
- C. Zou, Y. Gao, Y. Bin and Q. Zhai, Trans. Nonferrous Met. Soc. China, 2010, 20, 248–253 CrossRef CAS.
- M. K. Singh, M. C. Mathpal and A. Agarwal, Chem. Phys. Lett., 2012, 536, 87–91 CrossRef CAS.
- Z. Quan, Z. Luo, W. S. Loc, J. Zhang, Y. Wang, K. Yang, N. Porter, J. Lin, H. Wang and J. Fang, J. Am. Chem. Soc., 2011, 133, 17590–17593 CrossRef CAS PubMed.
- N. J. Greybush, V. Pacheco-Pena, N. Engheta, C. B. Murray and C. R. Kagan, ACS Nano, 2019, 13, 1617–1624 CrossRef CAS PubMed.
- X. Huang, J. Zhu, B. Ge, K. Deng, X. Wu, T. Xiao, T. Jiang, Z. Quan, Y. C. Cao and Z. Wang, J. Am. Chem. Soc., 2019, 141, 3198–3206 CrossRef CAS PubMed.
- Z. Quan, Z. Luo, Y. Wang, H. Xu, C. Wang, Z. Wang and J. Fang, Nano Lett., 2013, 13, 3729–3735 CrossRef CAS PubMed.
- S. H. Yu, A. Jin, X. Huang, Y. Yang, R. Huang, J. D. Brock, Y. E. Sung and H. D. Abruna, RSC Adv., 2018, 8, 23847–23853 RSC.
- J. Choi, S. Y. Han, J. Jin, J. Kim, J. H. Park, S. M. Lee, H. J. Kim and S. U. Son, J. Mater. Chem. A, 2013, 1, 8609–8615 RSC.
- X. Wang, X. Cao, L. Bourgeois, H. Guan, S. Chen, Y. Zhong, D. M. Tang, H. Li, T. Zhai, L. Li, Y. Bando and D. Golberg, Adv. Funct. Mater., 2012, 22, 2682–2690 CrossRef CAS.
- J. Chen, Y. Cheah, Y. Chen, N. Jayaprakash, S. Madhavi, Y. Yang and A. X. Lou, J. Phys. Chem. C, 2009, 113, 20504–20508 CrossRef CAS.
- Z. Quan, Y. Wang and J. Fang, Acc. Chem. Res., 2013, 46, 191–202 CrossRef CAS PubMed.
- N. S. Porter, H. Wu, Z. Quan and J. Fang, Acc. Chem. Res., 2013, 46, 1867–1877 CrossRef CAS PubMed.
- M. I. Bodnarchuk, K. V. Kravchyk, F. Krumeich, S. Wang and M. V. Kovalenko, ACS Nano, 2014, 8, 2360–2368 CrossRef CAS PubMed.
- K. Kravchyk, L. Protesescu, M. I. Bodnarchuk, F. Krumeich, M. Yarema, M. Walter, C. Guntlin and M. V. Kovalenko, J. Am. Chem. Soc., 2013, 135, 4199–4202 CrossRef CAS PubMed.
- Y. Idota, T. Kubota, A. Matsufuji, Y. Maekawa and T. Miyasaka, Science, 1997, 276, 1395 CrossRef CAS.
- B. Peng and J. Chen, Coord. Chem. Rev., 2009, 253, 2805–2813 CrossRef CAS.
- Y. Zhao, X. Li, B. Yan, D. Li, S. Lawes and X. Sun, J. Power Sources, 2015, 274, 869–884 CrossRef CAS.
- W. Sun, X. Rui, D. Yang, Z. Sun, B. Li, W. Zhang, Y. Zong, S. Madhavi, S. Dou and Q. Yan, ACS Nano, 2015, 9, 11371–11381 CrossRef CAS PubMed.
- H. Xie, W. P. Kalisvaart, B. C. Olsen, E. J. Luber, D. Mitlin and J. M. Buriak, J. Mater. Chem. A, 2017, 5, 9661–9670 RSC.
- B. Ji, F. Zhang, X. Song and Y. Tang, Adv. Mater., 2017, 29, 1700519 CrossRef PubMed.
- I. Sultana, T. Ramireddy, M. M. Rahman, Y. Chen and A. M. Glushenkov, Chem. Commun., 2016, 52, 9279–9282 RSC.
- H. Wang, Q. Wu, Y. Wang, X. Wang, L. Wu, S. Song and H. Zhang, Adv. Energy Mater., 2018, 9, 1802993 CrossRef.
- Z. Quan, D. Yang, P. Yang, X. Zhang, H. Lian, X. Liu and J. Lin, Inorg. Chem., 2008, 47, 9509–9517 CrossRef CAS PubMed.
- Z. Quan and J. Fang, Nano Today, 2010, 5, 390–411 CrossRef CAS.
- W. S. Loc, Z. Quan, C. Lin, J. Pan, Y. Wang, K. Yang, W. Jian, B. Zhao, H. Wang and J. Fang, Nanoscale, 2015, 7, 19047–19052 RSC.
- W. J. Baumgardner, Z. Quan, J. Fang and T. Hanrath, Nanoscale, 2012, 4, 3625–3628 RSC.
- Y. Nagaoka, R. Tan, R. Li, H. Zhu, D. Eggert, Y. A. Wu, Y. Liu, Z. Wang and O. Chen, Nature, 2018, 561, 378–382 CrossRef CAS PubMed.
- Y. Sun, X. Zuo, S. K. R. S. Sankaranarayanan, S. Peng, B. Narayanan and G. Kamath, Science, 2017, 356, 303–307 CrossRef CAS PubMed.
- X. X. Wang, S. Hwang, Y. T. Pan, K. Chen, Y. He, S. Karakalos, H. Zhang, J. S. Spendelow, D. Su and G. Wu, Nano Lett., 2018, 18, 4163–4171 CrossRef CAS PubMed.
- Z. Quan, D. Wu, J. Zhu, W. H. Everse, J. M. Boncella, L. D. A. Siebbeles, Z. Wang, A. Navrotsky and H. Xu, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9054–9057 CrossRef CAS.
- X. Song, S. Luo, X. Fan, M. Tang, X. Zhao, W. Chen, Q. Yang and Z. Quan, Front. Chem., 2018, 6, 468 CrossRef CAS PubMed.
- J. Zhu, Q. Di, X. Zhao, X. Wu, X. Fan, Q. Li, W. Song and Z. Quan, Inorg. Chem., 2018, 57, 6206–6209 CrossRef CAS PubMed.
- Z. Quan, W. Siu Loc, C. Lin, Z. Luo, K. Yang, Y. Wang, H. Wang, Z. Wang and J. Fang, Nano Lett., 2012, 12, 4409–4413 CrossRef CAS PubMed.
- D. V. Talapin, J. S. Lee, M. V. Kovalenko and E. V. Shevchenko, Chem. Rev., 2010, 110, 389–458 CrossRef CAS PubMed.
- S. Zhang, R. Geryak, J. Geldmeier, S. Kim and V. V. Tsukruk, Chem. Rev., 2017, 117, 12942–13038 CrossRef CAS PubMed.
- K. Kusada and H. Kitagawa, Adv. Mater., 2016, 28, 1129–1142 CrossRef CAS.
- Q. Yang, X. Zhao, X. Wu, M. Li, Q. Di, X. Fan, J. Zhu, X. Song, Q. Li and Z. Quan, Chem. Mater., 2019, 31, 2248–2252 CrossRef CAS.
- J. Zhang, H. Yang, B. Martens, Z. Luo, D. Xu, Y. Wang, S. Zou and J. Fang, Chem. Sci., 2012, 3, 3302–3306 RSC.
- J. Zhang and J. Fang, J. Am. Chem. Soc., 2009, 131, 18543–18547 CrossRef CAS PubMed.
- J. Zhang, Z. Luo, Z. Quan, Y. Wang, A. Kumbhar, D. M. Smilgies and J. Fang, Nano Lett., 2011, 11, 2912–2918 CrossRef CAS PubMed.
- X. Zhao, Q. Di, M. Li, Q. Yang, Z. Zhang, X. Guo, X. Fan, K. Deng, W. Chen, J. Zhang, J. Fang and Z. Quan, Chem. Mater., 2019 DOI:10.1021/acs.chemmater.9b00219.
- H. S. Im, Y. J. Cho, Y. R. Lim, C. S. Jung, D. M. Jang, J. Park, F. Shojaei and H. S. Kang, ACS Nano, 2013, 7, 11103–11111 CrossRef CAS PubMed.
- Y. Zhong, Z. Wang, R. Zhang, F. Bai, H. Wu, R. Haddad and H. Fan, ACS Nano, 2014, 8, 827–833 CrossRef CAS PubMed.
- M. Noh, Y. Kim, M. G. Kim, H. Lee, H. Kim, Y. Kwon, Y. Lee and J. Cho, Chem. Mater., 2005, 17, 3320–3324 CrossRef CAS.
- S. S. Chee and J. H. Lee, Electron. Mater. Lett., 2012, 8, 53–58 CrossRef CAS.
- L. Balan, R. Schneider, D. Billaud and J. Ghanbaja, Nanotechnology, 2005, 16, 1153–1158 CrossRef CAS.
- G. Saito, C. Zhu and T. Akiyama, Adv. Powder Technol., 2014, 25, 728–732 CrossRef CAS.
- L. Balan, R. Schneider, D. Billaud and J. Ghanbaja, Mater. Lett., 2005, 59, 1080–1084 CrossRef CAS.
- H. Bönnemann, W. Brijoux and T. Joussen, Angew. Chem., Int. Ed. Engl., 1990, 29, 273–275 CrossRef.
- C. Nayral, E. Viala, P. Fau, F. Senocq, J. C. Jumas, A. Maisonnat and B. Chaudret, Chem. – Eur. J., 2000, 6, 4082–4090 CrossRef CAS.
- S. Schlecht, M. Budde and L. Kienle, Inorg. Chem., 2002, 41, 6001–6005 CrossRef CAS PubMed.
- Y. Wang, J. Y. Lee and T. C. Deivaraj, J. Electrochem. Soc., 2004, 151, A1804–A1809 CrossRef CAS.
- Y. Wang, J. Y. Lee and T. C. Deivaraj, J. Mater. Chem., 2004, 14, 362–365 RSC.
- Y. Kwon, M. G. Kim, Y. Kim, Y. Lee and J. Cho, Electrochem. Solid-State Lett., 2006, 9, A34–A38 CrossRef CAS.
- B. Pejjai, V. R. Minnam Reddy, S. Gedi and C. Park, Int. J. Hydrogen Energy, 2017, 42, 2790–2831 CrossRef CAS.
- Y. C. Zhang, Z. N. Du, S. Y. Li and M. Zhang, Appl. Catal., B, 2010, 95, 153–159 CrossRef CAS.
- A. J. Biacchi, D. D. Vaughn and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 11634–11644 CrossRef CAS PubMed.
- M. D’Arienzo, D. Cristofori, R. Scotti and F. Morazzoni, Chem. Mater., 2013, 25, 3675–3686 CrossRef.
- M. S. Park, G. Wang, Y. Kang, D. Wexler, S. Dou and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 764–767 CrossRef.
- H. Zhang, Q. He, F. Wei, Y. Tan, Y. Jiang, G. Zheng, G. Ding and Z. Jiao, Mater. Lett., 2014, 120, 200–203 CrossRef CAS.
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS PubMed.
- W. Wang, M. Dahl and Y. Yin, Chem. Mater., 2013, 25, 1179–1189 CrossRef CAS.
- J. G. Railsback, A. C. Johnston-Peck, J. Wang and J. B. Tracy, ACS Nano, 2010, 4, 1913–1920 CrossRef CAS PubMed.
- H. Zeng, Curr. Nanosci., 2007, 3, 177–181 CrossRef CAS.
- F. Yang, L. Zhang, Z. Liu, S. Zhong, J. Ma and L. Bao, Adv. Mater. Sci. Eng., 2016, 2016, 15 Search PubMed.
- H. Ying and W. Han, Adv. Sci., 2017, 4, 1700298 CrossRef PubMed.
- Y. Chibane and M. Ferhat, J. Appl. Phys., 2010, 107, 053512 CrossRef.
- Z. Liu, G. S. Jackson and B. W. Eichhorn, Angew. Chem., Int. Ed., 2010, 122, 3241–3244 CrossRef.
- C. Dupont, Y. Jugnet and D. Loffreda, J. Am. Chem. Soc., 2006, 128, 9129–9136 CrossRef CAS PubMed.
- H. Li, Q. Wang, L. Shi, L. Chen and X. Huang, Chem. Mater., 2002, 14, 103–108 CrossRef CAS.
- L. Xiao, Y. Cao, J. Xiao, W. Wang, L. Kovarik, Z. Nie and J. Liu, Chem. Commun., 2012, 48, 3321–3323 RSC.
- P. Chen, L. Guo and Y. Wang, J. Power Sources, 2013, 222, 526–532 CrossRef CAS.
- D. D. Vaughn Ii and R. E. Schaak, Chem. Soc. Rev., 2013, 42, 2861–2879 RSC.
- K. Ramasamy, P. G. Kotula, A. F. Fidler, M. T. Brumbach, J. M. Pietryga and S. A. Ivanov, Chem. Mater., 2015, 27, 4640–4649 CrossRef CAS.
- R. J. Alan Esteves, S. Hafiz, D. O. Demchenko, Ü. Özgür and I. U. Arachchige, Chem. Commun., 2016, 52, 11665–11668 RSC.
- M. Sistani, M. S. Seifner, M. G. Bartmann, J. Smoliner, A. Lugstein and S. Barth, Nanoscale, 2018, 10, 19443–19449 RSC.
- M. S. Seifner, S. Hernandez, J. Bernardi, A. Romano-Rodriguez and S. Barth, Chem. Mater., 2017, 29, 9802–9813 CrossRef CAS.
- R. J. A. Esteves, M. Q. Ho and I. U. Arachchige, Chem. Mater., 2015, 27, 1559–1568 CrossRef CAS.
- X. Lu, K. J. Ziegler, A. Ghezelbash, K. P. Johnston and B. A. Korgel, Nano Lett., 2004, 4, 969–974 CrossRef CAS.
- D. H. Ha, T. Ly, J. M. Caron, H. Zhang, K. E. Fritz and R. D. Robinson, ACS Appl. Mater. Interfaces, 2015, 7, 25053–25060 CrossRef CAS PubMed.
- X. Xu, J. Zhuang and X. Wang, J. Am. Chem. Soc., 2008, 130, 12527–12535 CrossRef CAS PubMed.
- Z. Chen, X. Shi, L. Zhao and J. Zou, Prog. Mater. Sci., 2018, 97, 283–346 CrossRef CAS.
- L. Xu, C. Kim, A. K. Shukla, A. Dong, T. M. Mattox, D. J. Milliron and J. Cabana, Nano Lett., 2013, 13, 1800–1805 CrossRef CAS PubMed.
- H. Tabassum, R. Zou, A. Mahmood, Z. Liang, Q. Wang, H. Zhang, S. Gao, C. Qu, W. Guo and S. Guo, Adv. Mater., 2018, 30, 1705441 CrossRef PubMed.
- J. Zhang, H. Ren, J. Wang, J. Qi, R. Yu, D. Wang and Y. Liu, J. Mater. Chem. A, 2016, 4, 17673–17677 RSC.
- Y. J. Hong, M. Y. Son and Y. C. Kang, Adv. Mater., 2013, 25, 2279–2283 CrossRef CAS PubMed.
- M. Xie, X. Sun, S. M. George, C. Zhou, J. Lian and Y. Zhou, ACS Appl. Mater. Interfaces, 2015, 7, 27735–27742 CrossRef CAS.
- M. Mohamedi, S. J. Lee, D. Takahashi, M. Nishizawa, T. Itoh and I. Uchida, Electrochim. Acta, 2001, 46, 1161–1168 CrossRef CAS.
- J. Li, Y. Zhang, T. Gao, J. Han, X. Wang, B. Hultman, P. Xu, Z. Zhang, G. Wu and B. Song, J. Power Sources, 2018, 378, 105–111 CrossRef CAS.
- L. Yu, L. P. Wang, H. Liao, J. Wang, Z. Feng, O. Lev, J. S. C. Loo, M. T. Sougrati and Z. J. Xu, Small, 2018, 14, 1703338 CrossRef PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- Z. Li, J. Ding and D. Mitlin, Acc. Chem. Res., 2015, 48, 1657–1665 CrossRef CAS PubMed.
- M. Lao, Y. Zhang, W. Luo, Q. Yan, W. Sun and S. X. Dou, Adv. Mater., 2017, 29, 1700622 CrossRef PubMed.
- S. Yuan, Y. Zhu, W. Li, S. Wang, D. Xu, L. Li, Y. Zhang and X.-B. Zhang, Adv. Mater., 2017, 29, 1602469 CrossRef PubMed.
- W. Wang, P. Li, H. Zheng, Q. Liu, F. Lv, J. Wu, H. Wang and S. Guo, Small, 2017, 13, 1702228 CrossRef PubMed.
- Z. Zhang, X. Zhao and J. Li, Electrochim. Acta, 2015, 176, 1296–1301 CrossRef CAS.
- X. Wang, B. Liu, Q. Xiang, Q. Wang., X. Hou, D. Chen and G. Shen, ChemSusChem, 2014, 7, 308–313 CrossRef CAS PubMed.
- J. Hwang, S. Myung and Y. Sun, Adv. Funct. Mater., 2018, 28, 1802938 CrossRef.
- J. C. Pramudita, D. Sehrawat, D. Goonetilleke and N. Sharma, Adv. Energy Mater., 2017, 7, 1602911 CrossRef.
- I. Sultana, M. M. Rahman, Y. Chen and A. M. Glushenkov, Adv. Funct. Mater., 2018, 28, 1703857 CrossRef.
- W. Zhang, W. K. Pang, V. Sencadas and Z. Guo, Joule, 2018, 2, 1534–1547 CrossRef CAS.
- W. Zhang, Z. Wu, J. Zhang, G. Liu, N. H. Yang, R. S. Liu, W. K. Pang, W. Li and Z. Guo, Nano Energy, 2018, 53, 967–974 CrossRef CAS.
- J. Qian, Y. Xiong, Y. Cao, X. Ai and H. Yang, Nano Lett., 2014, 14, 1865–1869 CrossRef CAS.
- W. Zhang, J. Mao, S. Li, Z. Chen and Z. Guo, J. Am. Chem. Soc., 2017, 139, 3316–3319 CrossRef CAS.
- X. Zhao, W. Wang, Z. Hou, G. Wei, Y. Yu, J. Zhang and Z. Quan, Chem. Eng. J., 2019, 370, 677–683 CrossRef CAS.
- J. Qin, C. He, N. Zhao, Z. Wang, C. Shi, E. Liu and J. Li, ACS Nano, 2014, 8, 1728–1738 CrossRef CAS.
- Y. Xu, Q. Liu, Y. Zhu, Y. Liu, A. Langrock, M. R. Zachariah and C. Wang, Nano Lett., 2013, 13, 470–474 CrossRef CAS PubMed.
- J. Wang, W. Li, F. Wang, Y. Xia, A. M. Asiri and D. Zhao, Nanoscale, 2014, 6, 3217–3222 RSC.
- X. W. Lou, C. M. Li and L. A. Archer, Adv. Mater., 2009, 21, 2536–2539 CrossRef CAS.
- F. Zhang, J. Zhu, D. Zhang, U. Schwingenschlögl and H. N. Alshareef, Nano Lett., 2017, 17, 1302–1311 CrossRef CAS PubMed.
- Y. Zheng, T. Zhou, C. Zhang, J. Mao, H. Liu and Z. Guo, Angew. Chem., Int. Ed., 2016, 55, 3408–3413 CrossRef CAS PubMed.
- X. Xiong, C. Yang, G. Wang, Y. Lin, X. Ou, J. Wang, B. Zhao, M. Liu, Z. Lin and K. Huang, Energy Environ. Sci., 2017, 10, 1757–1763 RSC.
- Y. Jiang, M. Wei, J. Feng, Y. Ma and S. Xiong, Energy Environ. Sci., 2016, 9, 1430–1438 RSC.
- K. Huang, Z. Xing, L. Wang, X. Wu, W. Zhao, X. Qi, H. Wang and Z. Ju, J. Mater. Chem. A, 2018, 6, 434–442 RSC.
- Z. Huang, Z. Chen, S. Ding, C. Chen and M. Zhang, Solid State Ionics, 2018, 324, 267–275 CrossRef CAS.
- Q. Wu, Q. Shao, Q. Li, Q. Duan, Y. Li and H. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 15642–15651 CrossRef CAS.
- Z. Huang, Z. Chen, S. Ding, C. Chen and M. Zhang, Mater. Lett., 2018, 219, 19–22 CrossRef CAS.
- V. Lakshmi, Y. Chen, A. A. Mikhaylov, A. G. Medvedev, I. Sultana, M. M. Rahman, O. Lev, P. V. Prikhodchenko and A. M. Glushenkov, Chem. Commun., 2017, 53, 8272–8275 RSC.
| This journal is © The Royal Society of Chemistry 2019 |