
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative in situ13C NMR studies of the electro-catalytic oxidation of ethanol†
Jana Beatrice
Richter
a,
Claudia
Eßbach
b,
Irena
Senkovska
 b,
Stefan
Kaskel
b and
Eike
Brunner
*a
b,
Stefan
Kaskel
b and
Eike
Brunner
*a
aChair of Bioanalytical Chemistry, TU Dresden, Bergstraße 66, 01069 Dresden, Germany. E-mail: eike.brunner@tu-dresden.de
bChair of Inorganic Chemistry I, TU Dresden, Bergstraße 66, 01069 Dresden, Germany
First published on 30th April 2019
Abstract
The identification and quantification of reaction intermediates and products in a time-resolved fashion is of great value for the understanding of mechanisms underlying the electro-catalytic oxidation of alcohols. Using a newly developed in situ cell, the identification of different intermediates, products, and side products became possible by monitoring the ethanol electro-oxidation in operando by 13C NMR spectroscopy.
Electro-catalytic oxidation reactions of alcohols play a key role for future energy conversion applications. Increasing usage of renewable feedstock is essential since fossil fuels are limited and lead to the emission of greenhouse gas.1 Fuel cells are likely to play an important role for electro-mobility and other portable and stationary applications. Ethanol is a promising option as a hydrogen carrier because it is less toxic than other fuels and can be produced from biomass.2 Moreover, the selective electro-catalytic conversion of biomass-derived alcohols into aldehydes and other intermediates is a potential route towards fine chemicals.3–5 However, the mechanism of ethanol oxidation using platinum-based catalysts is not yet fully understood.6
Various spectro-electrochemical methods are already established in order to gain a comprehensive understanding of the mechanisms at the catalyst surface, e.g. IR- and Raman-spectroscopy, but also EPR and sXAS as well as invasive methods like MS.2,7–11In situ NMR spectroscopy has the following advantages: (i) the signal intensity for any detected species is directly proportional to the number of spins, i.e., NMR is fully quantitative. (ii) Small structural changes of molecules are sensitively monitored. (iii) No vacuum is required and the electrolyte does not perturb or contribute to the spectra.9,12 NMR spectroscopy can play a crucial role in the elucidation of reaction mechanisms because it allows to detect and quantify reaction intermediates and product molecules non-invasively in a time-resolved fashion and with high chemical specificity.13 Despite these advantages, combination of NMR spectroscopy and electrochemistry is still underdeveloped. Although NMR spectroscopy is a powerful method, its implementation in electrochemical studies is complicated by various technical issues. In comparison to other analytical methods, NMR spectroscopy has a relatively low sensitivity. Further problems occur for in situ studies because (a) metallic components of the cells cause problems especially if they are present in the detection coil and (b) interference between the current supplier and the spectrometer can amplify background noise.14 Thus, in situ monitoring of the reaction requires an appropriate experimental set-up. Metal electrodes for in situ investigations have to be radio frequency transparent, which depends on layer thickness and electrode size.15 Nevertheless, a few experimental set-ups have already been developed to analyse electrochemical reactions in situ by NMR. These experiments made use of interdigitated gold or epoxy-sealed, capillary-housed electrodes, which were inserted into a standard tube for solution NMR measurements.13,16–18
Metallic components of the electrochemical cell can be decreased by using carbon paper as electrode substrate. Such electrodes can be placed inside the coil, resulting in reduced line broadening in comparison to pure platinum or gold electrodes.17
In the present work, we describe a new cell design for in situ NMR investigations using coated carbon paper as electrodes in a sealed plastic bag cell placed inside the NMR detection coil. This set-up allows the in situ NMR detection of the electrode region where the reaction takes place. This enables direct monitoring of the reactions not only within the electrolyte but also on the catalytically active electrode. Another main advantage of the developed pouch cells is that they are compatible with any available solid-state NMR spectrometer. Additionally, the use of 13C-labeled educts provides a high sensitivity and allows to follow the reaction with a time resolution of the order of 10 minutes which is essential to understand the reaction mechanism.
The developed pouch cells are built in analogy to the plastic bag cells of BELLCORE which were used already to investigate electrochemical double-layer capacitors.19–21 They are variable in size and shape and can be measured easily within the 10 mm solenoid coil of the used probe. This set-up is illustrated in Fig. 1: scheme A shows all components of the pouch cell including working electrode (WE), counter electrode (CE), and separator film. Fig. 1B displays the used in situ probe with contacts for electrodes. The probe was manufactured by NMR Service GmbH, Erfurt, Germany. A pouch cell is shown in Fig. 1C.
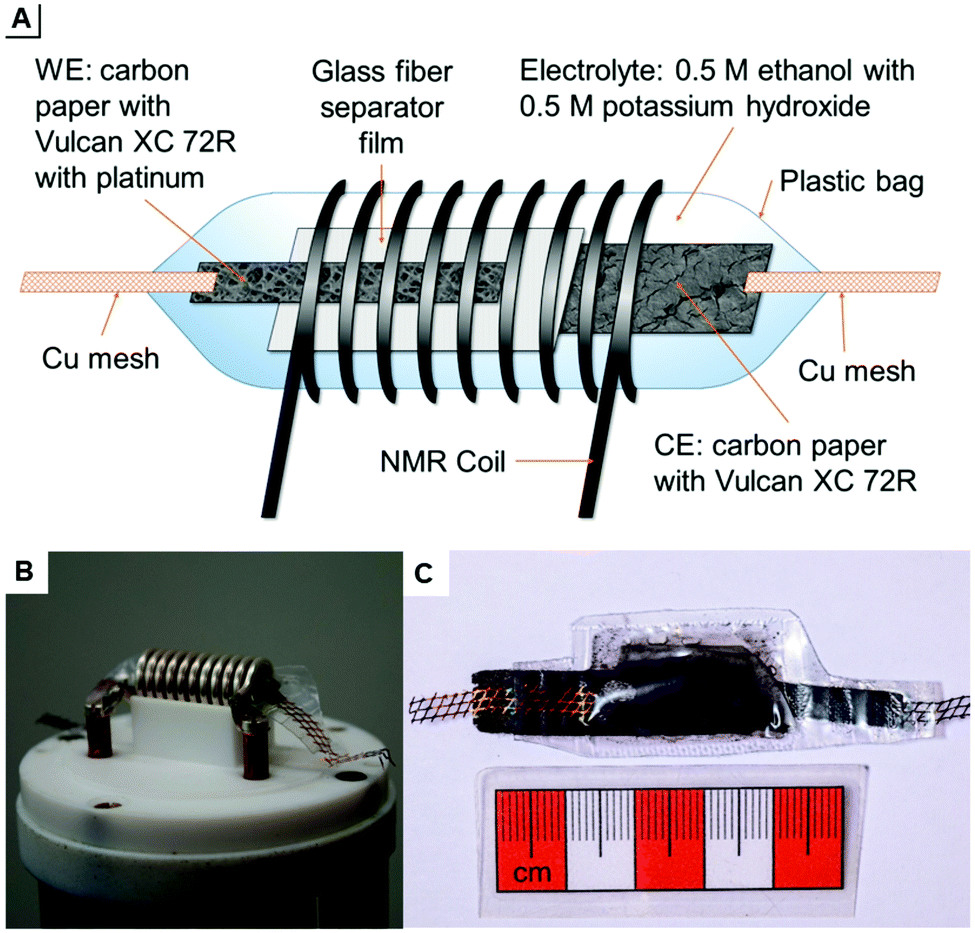 | ||
| Fig. 1 (A) Schematic drawing of the pouch cell for use in a 10 mm solenoid coil. (B) In situ NMR probe with a pouch cell inside the coil. To connect the cell with the probe, the copper mesh is pressed onto the contacts by a Teflon ring. (C) The pouch cell is about 4 cm long and 1 to 1.5 cm wide. | ||
The WE is based on carbon paper coated with the commercial electro-catalyst for ethanol oxidation, Pt/Vulcan XC 72R, exhibiting a total metal content of 10 wt%. The CE is also based on carbon paper coated with mesoporous carbon Vulcan XC 72R without Pt.
Coating was achieved by dispersing 100 mg of Pt/Vulcan XC 72R or Vulcan XC 72R in 1 mL ethanol and 1 mL deionized water for 5 min in a vibration ball mill, doctor blading on carbon paper (2 cm × 12 cm), air drying overnight, and activation at 80 °C in vacuum. Because of the instability and thickness of carbon paper, a copper mesh is used for contacting the electrodes with the probe and producing closed pouch cells. To avoid overpressure by carbon dioxide formation during the measurement, the closed pouch cell is opened with a needle on top immediately before the measurement. It should be noted that the reaction does not proceed in a fully sealed cell which is not opened prior to the measurement. After developing a certain gas pressure, the reaction does not further proceed (see ESI†). That means, electro-oxidation is inhibited in the presence of an overpressure due to carbon dioxide formation. Therefore, the cells were always perforated prior to the measurement in order to release CO2 overpressures. To avoid a short circuit, a glass fiber separator film is inserted between the two electrodes. The electrolyte consists of 0.5 M 13C-labeled ethanol and 0.5 M potassium hydroxide in water. All components of the pouch cell are sealed in a polypropylene plastic bag.
The NMR study is performed on a 300 MHz Bruker Avance spectrometer. To enhance the sensitivity, 13C-labeled ethanol CH313CH2OH is used. The in situ13C NMR spectra are acquired with single pulse excitation and 25 kHz spectral width. The 13C chemical shift is referenced relative to TMS. Fig. 2 shows spectra measured at 1.3 V over 100 hours reaction time. Additional measurements at other voltages are provided in the ESI.† The chronoamperometric current and the voltage were recorded during the experiment to monitor the stability of the operating cell (Fig. 3A). The practically constant chronoamperometric current indicates that the used in situ cells are indeed long-term stable. Due to technical and spatial limitations, it was not possible to integrate a reference electrode. However, the cell has a high resistance as indicated by the low current below 1 mA. Only minor deviations of the cell potential from the applied voltage are thus expected. The ethanol signal continuously decreases during the measurement time (cf.Fig. 2). Further signals are assigned to the reaction products acetic acid (177 ppm) and carbonate (160 ppm). Carbonate can be formed instead of carbon dioxide because the electrooxidation was carried out in alkaline medium. Within the reaction time, several signals of low intensity are constantly detected in subsequent spectra over certain time periods indicating intermediates or volatile products. These substances are acetaldehyde (208 ppm) which can be further oxidized to acetic acid, and carbon dioxide (120 ppm) which is volatile and evaporates from the cell or reacts with the electrolyte to carbonate. Additionally, a signal at ca. 90 ppm could be detected. It may result from ethane-1,1-diol (95 ppm) or ethoxyhydroxyethane (88 ppm). Both intermediates can be generated during the reaction of acetaldehyde with ethanol or water and were already found by Kim et al. in ex situ solution NMR studies.6
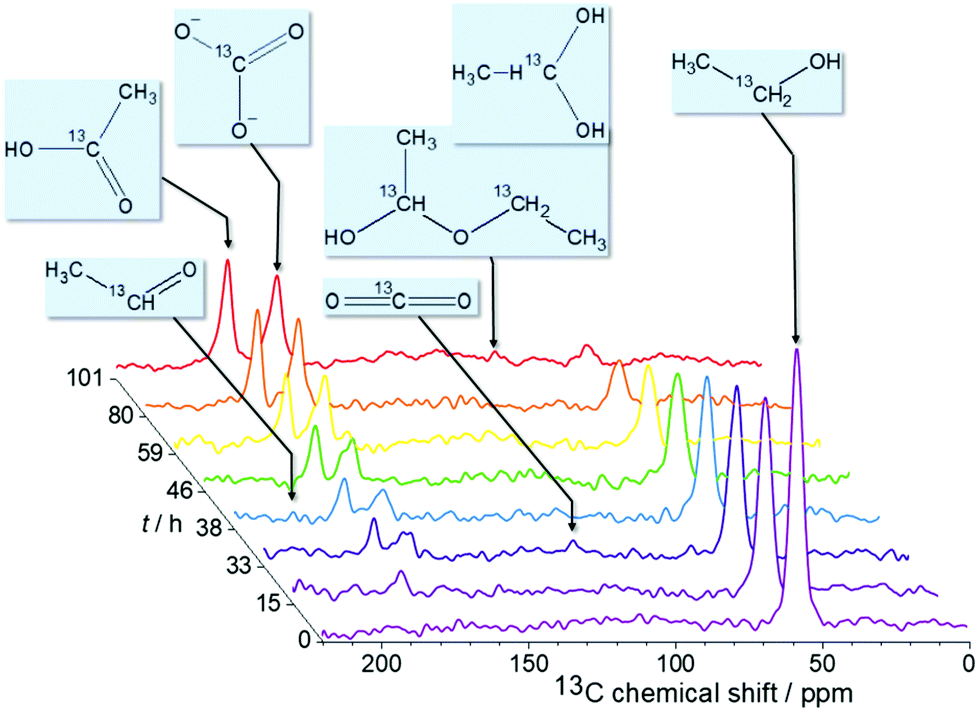 | ||
| Fig. 2 In situ 13C NMR spectra measured at 1.3 V as a function of reaction time t. The spectra show a conversion of ethanol and an increasing intensity of the signals due to acetic acid and carbonate. Weak intermediate signals could be assigned to acetaldehyde, carbon dioxide, as well as ethane-1,1-diol or ethoxyhydroxyethane. | ||
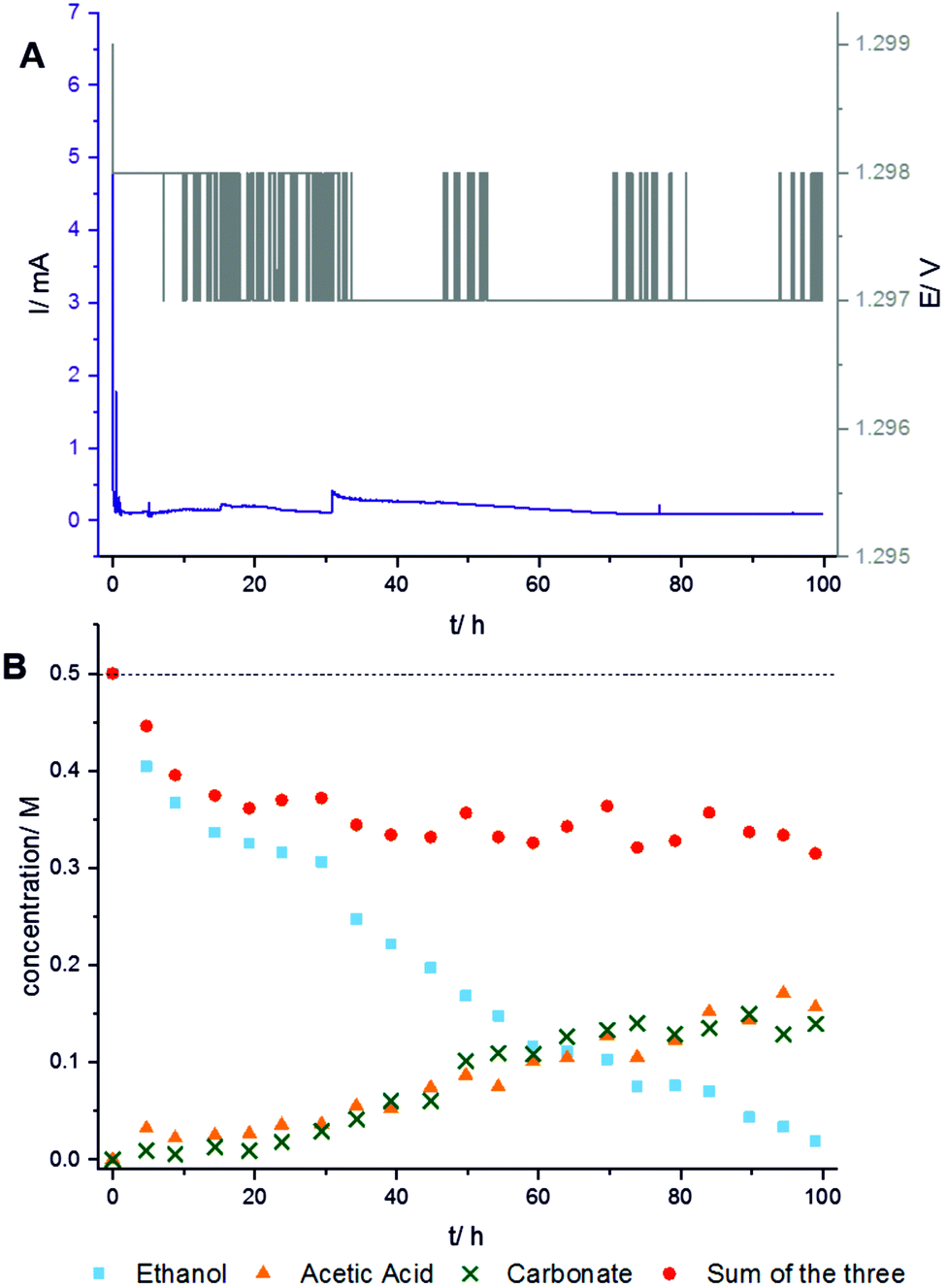 | ||
| Fig. 3 (A) Chronoamperometric current and applied voltage measured during the experiment shown in Fig. 2 (1.3 V over 100 hours). (B) Quantitative evaluation of the measurement reveals the conversion of ethanol (blue squares) into the main products acetic acid (orange triangles) and carbonate (green crosses). Red circles are the sum of the concentrations of ethanol, acetic acid, and carbonate. | ||
Quantification of the three most intense signals in the spectra measured at 1.3 V (Fig. 3B) shows the proceeding turnover of ethanol (blue rectangles) during the reaction time of 100 hours. The rapidly decreasing concentration of ethanol at practically constant current and voltage proves that the electro-oxidation reaction continuously proceeds. However, the concentration of carbonate (green crosses) and acetic acid (orange triangles) is low and increases only slowly within the first 20 hours. It is thus concluded that ethanol is mainly oxidized to volatile carbon dioxide which can escape from the cell. To determine the maximum possible amount of volatile carbon dioxide, the sum of the measured concentrations of ethanol, acetic acid, and carbonate (red points) must be subtracted from the total initial concentration (horizontal dashed line). Indeed, a rapidly growing difference is observed in the first 20 hours. After reaction times between 20 and 60 hours, the concentrations of both, acetic acid and carbonate, strongly increase. The sum of the concentrations of ethanol, acetic acid, and carbonate (red points) remains then nearly constant meaning that almost no volatile carbon dioxide is formed for reaction times longer than 20 hours. Carbon dioxide then obviously reacts to carbonate. It is possible that some carbonate is initially adsorbed at the catalyst surface. This carbonate would not be detectable due to line broadening caused by immobilization. Strong adsorption of carbonate at the catalyst may also lead to slow product release into the solution. Thus, carbonate formed at the beginning would be detected later.22 Therefore, the reaction rate for the oxidation to carbon dioxide may be limited by the decreased rate of C–C-bond cleavage since the catalytic surface would be partly blocked by poisoning species like carbonate.23
In summary, the developed spectro-electrochemical NMR technique using customized pouch cells provides a simple and robust method to analyse product spectra of electro-catalytic reactions in operando. The use of 13C-labeled educts provides a good signal-to-noise ratio as well as time resolution due to the short measurement time. The long-term stable pouch cell set-up allows to follow the reaction until complete ethanol conversion. Acetic acid and carbonate could be quantified as main products. In addition, four different intermediates of the oxidation reaction mechanism are detected. This in situ method is a valuable tool for future investigations such as the development of electro-catalysts with higher selectivity towards specific alcohols. It allows to analyse the influence of relevant reaction parameters like voltage, educt concentration etc. for achieving higher selectivity and elucidation of electro-oxidation mechanisms. The developed set-up is also applicable to study other electro-catalytic reactions.
Financial support by the DFG (Priority Program 1928 “COORNET”) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Blanc, M. Leskes and C. P. Grey, Acc. Chem. Res., 2013, 46, 1952 CrossRef CAS PubMed.
- F. Vigier, C. Coutanceau, F. Hahn, E. M. Belgsir and C. Lamy, J. Electroanal. Chem., 2004, 563, 81 CrossRef CAS.
- R. Ciriminna, V. Pandarus, F. Béland, Y. J. Xu and M. Pagliaro, Org. Process Res. Dev., 2015, 19, 1554 CrossRef CAS.
- Y. Kwon, Y. Birdja, I. Spanos, P. Rodrguez and M. T. M. Koper, ACS Catal., 2012, 2, 799 Search PubMed.
- H. Liu, J. Qin, S. Zhao, Z. Gao, Q. Fu and Y. Song, Electrochem. Commun., 2019, 98, 53 CrossRef CAS.
- I. Kim, O. H. Han, S. A. Chae, Y. Paik, S.-H. Kwon, K.-S. Lee, Y.-E. Sung and H. Kim, Angew. Chem., Int. Ed., 2011, 50, 2270 CrossRef CAS PubMed.
- M. Zhang, M. de Respinis and H. Frei, Nat. Chem., 2014, 6, 362 CrossRef CAS PubMed.
- C. Gimbert-Surinach, D. Moonshiram, L. Francás, N. Planas, V. Bernales, F. Bozoglian, A. Guda, L. Mognon, I. López, M. A. Hoque, L. Gagliardi and C. J. Cramer, J. Am. Chem. Soc., 2016, 138, 15291 CrossRef CAS PubMed.
- S. Klod and L. Dunsch, Magn. Reson. Chem., 2011, 49, 725 CrossRef CAS PubMed.
- Z. Cui, C. Xie, X. Feng, N. Becknell, P. Yang, Y. Lu, X. Zhai, X. Liu, W. Yang, Y.-D. Chuang and J. Guo, J. Phys. Chem. Lett., 2017, 8, 319 CrossRef CAS PubMed.
- A. A. Abd-El-Latif, E. Mostafa, S. Huxter, G. Attard and H. Baltruschat, Electrochim. Acta, 2010, 55, 7951 CrossRef CAS.
- X. Liu, D. Wang, G. Liu, V. Srinivasan, Z. Liu, Z. Hussain and W. Yang, Nat. Commun., 2013, 4, 2568 CrossRef.
- E. G. Sorte and Y. Y. J. Tong, J. Electroanal. Chem., 2016, 769, 1 CrossRef CAS.
- F. Poli, J. S. Kshetrimayum, L. Monconduit and M. Letellier, Electrochem. Commun., 2011, 13, 1293 CrossRef CAS.
- S. Klod, F. Ziegs and L. Dunsch, Anal. Chem., 2009, 81, 10262 CrossRef CAS.
- E. G. Sorte, S. Jilani and Y. Y. J. Tong, Electrocatalysis, 2017, 8, 95 CrossRef CAS.
- L. M. S. Nunes, T. B. Moraes, L. L. Barbosa, L. H. Mazo and L. A. Colnago, Anal. Chim. Acta, 2014, 850, 1 CrossRef CAS PubMed.
- L. Huang, E. G. Sorte, S.-G. Sun and Y. Y. J. Tong, Chem. Commun., 2015, 51, 8086 RSC.
- J.-M. Tarascon, A. S. Gozdz, C. Schmutz, F. Shokoohi and P. C. Warren, Solid State Ionics, 1996, 86–88, 49 CrossRef CAS.
- H. Wang, T. K.-J. Köster, N. M. Trease, J. Ségalini, P.-L. Taberna, P. Simon, Y. Gogotsi and C. P. Grey, J. Am. Chem. Soc., 2011, 133, 19270 CrossRef CAS PubMed.
- A. J. Ilott, N. M. Trease, C. P. Grey and A. Jerschow, Nat. Commun., 2014, 5, 4536 CrossRef CAS.
- J. S. Spendelow and A. Wieckowski, Phys. Chem. Chem. Phys., 2007, 9, 2654 RSC.
- G. García, P. Rodríguez, V. Rosca and M. T. M. Koper, Langmuir, 2009, 25, 13661 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc02660f |
| This journal is © The Royal Society of Chemistry 2019 |