
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oriented attachment of VNAR proteins, via site-selective modification, on PLGA–PEG nanoparticles enhances nanoconjugate performance†
João C. F.
Nogueira
a,
Michelle K.
Greene
b,
Daniel A.
Richards
ac,
Alexander O.
Furby
a,
John
Steven
de,
Andrew
Porter
de,
Caroline
Barelle
*de,
Christopher J.
Scott
 *b and
Vijay
Chudasama
*af
*b and
Vijay
Chudasama
*af
aDepartment of Chemistry, University College London, London, UK. E-mail: v.chudasama@ucl.ac.uk
bCentre for Cancer Research and Cell Biology, School of Medicine, Queen's University Belfast, Belfast, UK. E-mail: c.scott@qub.ac.uk
cDepartment of Materials, Imperial College London, London, UK
dInstitute of Medical Science, University of Aberdeen, Aberdeen, UK
eElasmogen Ltd, Aberdeen, UK. E-mail: caroline.barelle@elasmogen.com
fResearch Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal
First published on 29th May 2019
Abstract
Herein we report the construction of a nanoparticle-based drug delivery system which targets a key regulator in tumour angiogenesis. We exploit a Variable New Antigen Receptor (VNAR) domain, conjugated using site-specific chemistry, to direct poly lactic acid-co-glycolic acid–polyethylene glycol (PLGA–PEG) nanoparticles to delta like canonical Notch ligand 4 (DLL4). The importance of site-specific chemistry is demonstrated.
Over the last few years the utilisation of nanotechnology within biomedicine, particularly in the field of targeted therapy, has grown considerably.1,2 Nanoscale materials can cross biological barriers including the blood–brain barrier,3 transit in and out of blood vessels4 or even passively penetrate the cell membrane by different mechanisms (e.g. endocytosis).5 Furthermore, nanoparticles (NPs) are able to safely transport different types of cargo, such as molecular imaging agents or cytotoxic drugs in high quantities.6 Most recently, NPs have also been combined with targeting proteins, such as antibodies or antibody fragments, to enable the targeting of disease biomarkers in a concept tagged “active targeting”.7 Although antibodies have been employed for the generation of actively targeted NPs, the clinical application of the resultant nanoconjugates has remained rare. This is due in part to suboptimal methods for the appendage of proteins to NPs.8 For instance, many protocols employ random conjugation methods, such as electrostatic adsorption of proteins to NP surfaces, to achieve targeting.9 This method is mediated by the surface charge of antibodies, which varies significantly from antibody to antibody, and can lead to significant batch-to-batch variability of resulting nanoconjugates and poor stability.10,11 To overcome these issues, methods which rely on chemically stable covalent bonds to graft antibodies to NPs have been developed; these methods are reported to confer better stability in vivo.12 Early developers focused on covalently attaching antibodies (via surface lysine residues) to NP via carbodiimide chemistry.13–15 However, this is sub-optimal as it usually presents low reaction efficiencies in aqueous conditions due to competing hydrolysis.16 Conjointly, the modification of lysine residues affords very little control over the orientation of the antibody targeting ligands on the NP surface, limiting antigen-binding and overall target avidity. We recently demonstrated the importance of controlled chemical ligation for successful nanoconjugate performance, particularly in the context of target affinity.17a This study showed that using site-selective chemistry to conjugate Trastuzumab antibody fragments (i.e. F(ab)s) to PLGA–PEG NPs resulted in superior antigen binding when compared to using classical lysine-based approaches.17a
There is a growing trend in NP construction towards utilising small antibody fragments rather than full antibodies as active targeting ligands.18 Smaller size permits a greater loading on NP surfaces, whilst maintaining the antigen-binding capability of a full antibody, and absence of a fragment crystallisable (Fc) region can reduce Fc-mediated immunogenicity.18–20 Methods of generating antibody fragments are well described in literature, e.g. F(ab)′ and F(ab) (both ca. 50 kDa) can be generated through enzymatic digestions,18 and have been successfully employed in the formation of nanoconjugates for the treatment of multiple diseases.17a,21,22 However, there is a drive to find alternative proteins that share the same specific binding properties of antibodies but that are of an even smaller size. Shark Variable New Antigen Receptors (VNARs), at only ca. 11 kDa, are seen as an attractive alternative to current traditional antibodies and their fragments as they hold many advantages such as higher stability, solubility and low cost of manufacturing, while upholding high specificity to bind to novel or cryptic epitopes.23–25 Most relevantly, the simple VNAR molecular architecture presents a versatile platform for re-formatting and engineering. For instance, VNARs have been previously engineered to increase their solubility and refolding ability26 and also to bind to many different antigens with high specificity.27 Also, VNARs are reported to tolerate extreme pH values (down to pH 1.5) and high temperature conditions. These advantages have allowed the facile generation of a plethora of new biologics.28 However, their applications within nanotechnology remain to be exploited.
Herein is presented a novel PLGA–PEG NP–VNAR conjugate that targets the inhibition of endothelial sprouting and proliferation (processes involved in tumour angiogenesis).29 We hypothesised that by applying a highly-directed chemical strategy for decorating nanocarriers with VNAR ligands, the overall potential of VNAR directed nanocarriers could be maximised, i.e. antigen binding enhancement due to the favourable orientation and large number of proteins packed on the NP surface. To ensure modularity, we created a platform that is amenable to commonly employed and well-tolerated copper (Cu)-free “click” chemistry, namely strain-promoted azide–alkyne cycloaddition (SPAAC) chemistry.
PLGA–PEG polymeric NPs were chosen as the nanocarrier platform due to their well-established ability to effectively hold cargo for controlled drug delivery.17a However, we anticipate that the findings of this study could be extrapolated to emerging nanocarriers e.g. poly(ε-caprolactone)–PEG, which recent findings have suggested could be ideal drug carriers due to excellent cellular internalization and prolonged blood circulation time.17b,c Owing to our past experience in synthesizing such PLGA–PEG constructs we used a 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 blend of PLGA-502H:PLGA–PEG-azide to formulate our NP-azide constructs for “click” modification, as these NPs exhibit long-term stability.17a
:25 blend of PLGA-502H:PLGA–PEG-azide to formulate our NP-azide constructs for “click” modification, as these NPs exhibit long-term stability.17a
Subsequently, an anti-DLL4 E4 VNAR clone, hereafter referred to as VNAR E4 6, was used as a model VNAR targeting moiety. VNAR E4 6 clone was specifically expressed to recognise and bind to DLL4, a key regulator that activates Notch signaling pathways directly related with early embryonic vascular development in tumor angiogenesis (see ESI,† for details).30 An Alanine–Cysteine–Alanine (ACA) sequence was inserted at the C-terminal region, enabling site-selective cysteine modification. Also, this reactive cysteine thiol handle was inserted distal from the antigen binding site, at the N-terminus, in view of maximising orientation benefits and minimising any disturbance of the paratope–epitope interaction. To site-selectively modify VNAR E4 6 and introduce a strained alkyne moiety we synthesised monobromopyridazinedione 5. Bromopyridazinediones are selectively reactive towards cysteine-bearing proteins, and bicyclo[6.1.0]nonyne (BCN(endo)) (BCN) is well suited to “click” reactions with azide-bearing NPs in aqueous buffers.31 Synthesis of strained alkyne monobromopyridazinedione (PD 5) proceeded from readily available starting materials in a facile manner over five steps (Scheme 1).32 Initially, di-Boc protection of methyl-hydrazine 1 and subsequent Michael addition to tert-butyl acrylate yielded hydrazine 2. Following this, deprotection and dehydration under acidic conditions afforded acid PD 3. Subsequent esterification of acid 3 with NHS afforded amine-reactive PD 4. In the final step, an amide coupling between commercially available BCN(endo)–PEG2–NH2 and PD 4 yielded the desired heterobifunctional PD linker 5 (Scheme 1).
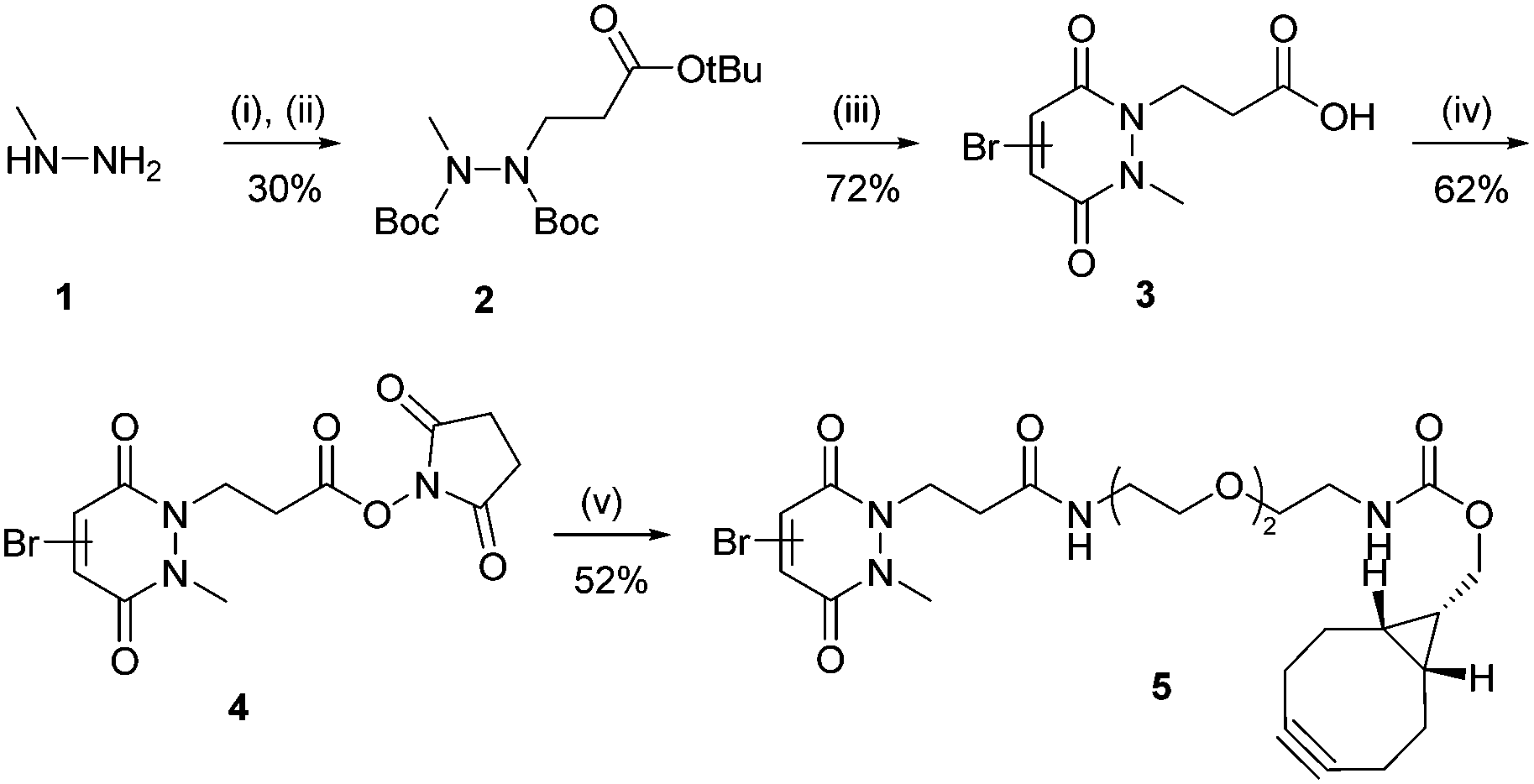 | ||
| Scheme 1 Synthesis route of PD 5. Reagents and conditions: (i) Boc anhydride, i-PrOH, DCM, 21 °C, 16 h; (ii) tert-butyl acrylate, i-PrOH, 60 °C, 24 h; (iii) bromomaleic acid, AcOH, reflux, 5 h; (iv) DCC, NHS, THF, 21 °C, 16 h, (v) BCN(endo)–PEG2–NH2, MeCN, 21 °C, 16 h. | ||
Bioconjugation of native VNAR E4 6 with PD 5 was appraised via SDS-PAGE and LC-MS, where it was observed that complete conversion only occurred following pre-reduction of VNAR E4 6 with tris(2-carboxyethyl)phosphine (TCEP). This can be explained by the fact that the single cysteine can be capped with glutathione or even spontaneously form a disulfide dimer in solution. Several optimisation parameters were tuned to obtain pure VNAR conjugate 7 (see ESI,† for details). The optimised conditions for successful site-selective modification comprised pre-incubation of VNAR E4 6 with TCEP (10 eq.), followed by addition of PD 5 (20 eq.); full reduction and complete conversion were confirmed by LC-MS. To demonstrate the availability of VNAR conjugate 7 to participate in a “click” reaction, the construct was successfully reacted with Alexafluor®-488-N3 (Scheme 2, conjugate 8). Having successfully modified the C-terminal cysteine residue of the VNAR E4 with PD 5, we next aimed to couple the protein to the surface of polymeric azide NPs in a site-selective manner. Previously, we have shown that similar heterobifunctional linkers can be introduced into antibody F(ab) fragments, thereby facilitating site-specific “click” conjugation to complementary azide-functionalised PLGA–PEG NPs of approximately 200 nm in diameter (Table S1, ESI†).17a In concert with this strategy, the strained alkyne functionality of VNAR conjugate 7 was reacted with surface-exposed azide groups on PLGA–PEG-azide NP 10via Cu-free SPAAC chemistry to generate E4 functionalised NP (site-selective cysteine modified E4 NP 12) of 214.2 ± 13.3 nm in size (Fig. 1 and Table S1, ESI†). A benchmark formulation was also included in this study to allow direct comparison of our coupling strategy with carbodiimide chemistry; an established nanoconjugation approach that has been widely implemented in the development of targeted NP modules. Here, amine groups distributed throughout native VNAR E4 6 (i.e. containing no PD 5 linker) were reacted with NHS esters on the surface of PLGA–PEG–NHS NP 9, with coupling proceeding via amide bond formation to generate a nanoconjugate of 197.8 ± 5.1 nm in size (random lysine modified E4 NP 11). Interestingly, the conjugation efficiency was revealed to be greater for the SPAAC click chemistry when compared to the control; this is in agreement with our previously reported data.17a Also, non-targeted NHS and azide NP controls were utilised, of 191.5 ± 5.3 nm and 194.0 ± 2.5 nm in size, respectively (NHS NP 9 and azide NP 10). To confirm that VNAR E4 still retained its antigen binding affinity after chemical manipulation and nanoconjugation, fluorescently labelled NPs conjugated to VNAR E4, i.e.11 and 12, were incubated with recombinant human DLL4 immobilised on microtiter plates. Dose-dependent binding of NP 12 to DLL4 was observed, with each increment in NP concentration leading to a stepwise enhancement in fluorescence (Fig. 2). Despite similar concentration-dependent binding of NP 11 to DLL4, fluorescence readouts were significantly lower than those observed for NP 12. The controls showed the binding of non-targeted control NPs (i.e. NPs 9 and 10) to be negligible. Moreover, to confirm that the enhanced binding of NP 12 was not simply due to the higher protein loading on these NP, we next formulated NP 11 and NP 12 to present equal amounts of VNAR E4 6 and VNAR conjugate 7, respectively, on their surfaces. Again, DLL4 binding by NP 12 remained superior (Fig. S3, ESI†). Collectively, these findings demonstrate that our novel chemistry may be exploited for the site-specific “click” conjugation of DLL4-targeted VNARs to polymeric NPs, yielding nanoconjugates with superior binding ability than those formulated using conventional methods.
 | ||
| Scheme 2 Bioconjugation of VNAR E4 6 with PD 5 and subsequent “click” reaction with Alexafluor®-488-N3. Reagents and conditions: (vi) TCEP·HCl (10 eq.), PD 5 (20 eq.), phosphate buffer pH 7.4, 21 °C, 16 h. (vii) Alexafluor®-488-N3, phosphate buffer pH 7.4, 21 °C, 5 h. | ||
 | ||
| Fig. 1 Representation of all nanoformulations tested: NHS NP 9; azide NP 10; random lysine modified E4 NP 11 and site-selective cysteine modified E4 NP 12. | ||
 | ||
| Fig. 2 Binding of site-selective cysteine modified E4 NP 12 to DLL4 is greatly enhanced compared to random lysine modified E4 NP 11. Binding of fluorescently labelled NPs 11 & 12, and corresponding blank NP controls 9 & 10 (125, 250 and 500 μg polymer mL−1) to DLL4 was analysed by modified ELISA. Data expressed as mean ± SEM. | ||
Following this, we sought to verify that the observed enhancement in fluorescence in the above binding assays was attributed to specific interactions between the targeted nanoformulations and the immobilised DLL4 antigen. To do this, the E4 paratopes on the surface of the NP were saturated with an excess of free DLL4 prior to incubation in microtiter plate wells coated with the same antigen. Binding of NP 11 and 12 was inhibited following pre-incubation with DLL4, as evidenced by significantly lower fluorescence readouts for these samples (Fig. 3A). As an alternative approach, both NP 12 and an anti-DLL4 monoclonal antibody were added simultaneously to DLL4-immobilised wells. In these studies, NP binding was progressively impeded with increasing concentrations of competing anti-DLL4 (Fig. 3B). Taken together, these experiments provide robust confirmation that the ability of the NP formulations to bind to DLL4 was dependent upon the surface conjugation of the VNAR proteins.
 | ||
| Fig. 3 Binding of E4 nanoformulations is DLL4-specific. (A) Binding of fluorescently labelled NP 11, NP 12 and corresponding blank NP controls 9 and 10 (50 μg polymer mL−1) to DLL4 was analysed by modified ELISA ± pre-incubation with DLL4 (10 μg mL−1). (B) Binding of fluorescently labelled NP 12 (250 μg polymer mL−1) to DLL4 was analysed by modified ELISA ± competition with DLL4 antibody (165–40 000 ng mL−1). (C) Enhanced DLL4 binding by NP 12 is dependent upon site-specific “click” coupling. VNAR conjugate 7 and VNAR E4 6 were incubated with fluorescently labelled NP composed of (1) PLGA-502H, (2) a 75:25 blend of PLGA-502H:PLGA-PEG-NHS or (3) a 75:25 blend of PLGA-502H : PLGA-PEG-azide. Binding of these nanoformulations and corresponding blank NP controls (500 μg polymer mL−1) to DLL4 was analysed by modified ELISA. Data expressed as mean ± SEM. | ||
We next investigated whether the superior binding of site-selective cysteine modified E4 NP 12 was contingent upon both surface display of azide and cysteine modification of the VNAR. Various nanoformulations were synthesised by incubating strained alkyne bearing VNAR conjugate 7 with NP comprised solely of PLGA-502H, or a blend of PLGA-502H and either PLGA–PEG–NHS or PLGA–PEG-azide. Binding of these NP to immobilised DLL4 was minimal, except for those formulated via “click” coupling of VNAR conjugate 7 to complementary azide-terminated NP (Fig. 3C). Furthermore, native VNAR E4 6 was also incubated with the above polymeric NP formulations; all three formulations showed only marginal levels of DLL4 binding (Fig. 3C). These findings clearly indicate that the enhanced DLL4 binding activity of NP 12 is not simply mediated via non-specific surface adsorption of the VNAR clone. Rather it shows that the presence of the surface-exposed azide and the strained-alkyne modified VNAR are critical determinants of nanoconjugate performance. This confirms the site-selectivity and importance of covalent conjugation.
As a final set of studies, we explored the impact of different loadings of strained alkyne bearing VNAR conjugate 7 on the surface of the NP. As expected, incremental addition of VNAR conjugate 7 resulted in cumulative improvements in DLL4 binding. Interestingly, fluorescence levels plateaued upon adding >2 nanomoles of VNAR conjugate 7 per milligram of polymer, suggesting epitope saturation (see Fig. S4, ESI†).
In conclusion, this study showcases a new modular method for attaching VNAR proteins onto the surfaces of PLGA–PEG polymeric NPs via “click” chemistry. A novel heterobifunctional PD linker was designed to enable site-selective cysteine modification of a VNAR clone that was attached to an azide decorated NP via SPAAC conjugation. This NP–VNAR construct, with orientated protein presentation on the NP surface, showed favourable properties in terms of target binding when compared to traditional NP–protein conjugation chemistries.
This work was partially funded through a US-Ireland R&D Partnership grant (STL/5010/14, MRC grant MC_PC_15013). JCFN is funded by the EU's Horizon 2020 programme under Marie-Curie grant agreement 675007. We acknowledge UCL Chemistry Mass Spectrometry Facility (Dr K. Karu/Dr X. Yang).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Wang and M. Thanou, Pharmacol. Res., 2010, 62, 90–99 CrossRef CAS PubMed; (b) L. Cai, Z. Gu, J. Zhong, D. Wen, G. Chen, L. He, J. Wu and Z. Gu, Drug Discovery Today, 2018, 23, 1126–1138 CrossRef CAS PubMed.
- O. Salata, J. Nanobiotechnol., 2004, 2, 3 CrossRef PubMed.
- C. Saraiva, C. Praça, R. Ferreira, T. Santos, L. Ferreira and L. Bernardino, J. Controlled Release, 2016, 235, 34–47 CrossRef CAS PubMed.
- D. Jiang, M. Niwa and A. C. Koong, Semin. Cancer Biol., 2015, 33, 48–56 CrossRef CAS PubMed.
- T. G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- F. U. Din, et al. , Int. J. Nanomed., 2017, 12, 7291–7309 CrossRef PubMed.
- R. Solaro, F. Chiellini and A. Battisti, Materials, 2010, 3, 1928–1980 Search PubMed.
- M. Mahmoudi, Nat. Nanotechnol., 2018, 13, 775–776 CrossRef CAS PubMed.
- A. J. Sivaram, A. Wardiana, C. B. Howard, S. M. Mahler and K. J. Thurecht, Adv. Healthcare Mater., 2018, 7, 1700607 CrossRef PubMed.
- R. G. Couston, M. W. Skoda, S. Uddin and C. F. van der Walle, mAbs, 2013, 5, 126–139 CrossRef PubMed.
- Y. Li, X. Chen and N. Gu, J. Phys. Chem. B, 2008, 112, 16647–16653 CrossRef CAS PubMed.
- M. Arruebo, M. Valladares and Á. González-Fernández, J. Nanomater., 2009, 1–24 Search PubMed.
- J. Xiong, S. Han, S. Ding, J. He and H. Zhang, Oncol. Rep., 2018, 39, 1396–1404 CAS.
- S. K. Vashist, Diagnostics, 2012, 2, 23–33 CrossRef CAS PubMed.
- D. Bartczak and A. G. Kanaras, Langmuir, 2011, 27, 10119–10123 CrossRef CAS PubMed.
- E. R. Figueroa, A. Y. Lin, J. Yan, L. Luo, A. E. Foster and R. A. Drezek, Biomaterials, 2014, 35, 1725–1734 CrossRef CAS PubMed.
- (a) M. K. Greene, D. A. Richards, J. C. F. Nogueira, K. Campbell, P. Smyth, M. Fernández, C. J. Scott and V. Chudasama, Chem. Sci., 2018, 9, 79–87 RSC; (b) Y. Niu, F. J. Stadler, T. He, X. Zhang, Y. Yu and S. Chen, J. Mater. Chem. B, 2017, 5, 9477–9481 RSC; (c) Y. Niu, T. He, J. Song, S. Chen, X. Liu, Z. Chen, Y. Yu and S. Chen, Chem. Commun., 2017, 53, 7541–7544 RSC.
- D. A. Richards, A. Maruani and V. Chudasama, Chem. Sci., 2017, 8, 63–77 RSC.
- G. A. Pietersz, X. Wang, M. L. Yap, B. Lim and K. Peter, Nanomedicine, 2017, 12, 1873–1889 CrossRef CAS PubMed.
- D. A. Richards, Drug Discovery Today: Technol., 2018, 30, 35–46 CrossRef PubMed.
- S. Chen, S. Florinas, A. Teitgen, Z.-Q. Xu, C. Gao, H. Wu, K. Kataoka, H. Cabral and R. J. Christie, Sci. Technol. Adv. Mater., 2017, 18, 666–680 CrossRef CAS.
- W. W. K. Cheng and T. M. Allen, J. Controlled Release, 2008, 126, 50–58 CrossRef CAS PubMed.
- C. Barelle and A. Porter, Antibodies, 2015, 4, 240–258 CrossRef CAS.
- M. Kovaleva, L. Ferguson, J. Steven, A. Porter and C. Barelle, Expert Opin. Biol. Ther., 2014, 14, 1527–1539 CrossRef CAS PubMed.
- O. C. Ubah, et al. , Biochem. Soc. Trans., 2018, 46, 1559–1565 CrossRef CAS PubMed.
- E. R. Goldman, J. L. Liu, D. Zabetakis and G. P. Anderson, Front. Immunol., 2017, 8, 865 CrossRef PubMed.
- H. Dooley, M. F. Flajnik and A. J. Porter, Mol. Immunol., 2003, 40, 25–33 CrossRef CAS PubMed.
- O. C. Ubah, J. Steven, M. Kovaleva, L. Ferguson, C. Barelle, A. J. R. Porter and C. J. Barelle, Front. Immunol., 2017, 8, 1780 CrossRef PubMed.
- M. E. Pitulescu, et al. , Nat. Cell Biol., 2017, 19, 915–927 CrossRef CAS PubMed.
- T. Yoneya, T. Tahara, K. Nagao, Y. Yamada, T. Yamamoto, M. Osawa, S. Miyatani and M. Nishikawa, J. Biochem., 2001, 129, 27–34 CrossRef CAS PubMed.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
- C. Bahou, D. A. Richards, A. Maruani, E. A. Love, F. Javaid, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2018, 16, 1359–1366 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc02655j |
| This journal is © The Royal Society of Chemistry 2019 |