
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Buchwald Hartwig diversification of unprotected halotryptophans, halotryptophan containing tripeptides and the natural product barettin in aqueous conditions†
Yohann J. G.
Renault‡
a,
Rosemary
Lynch‡
a,
Enrico
Marelli‡
a,
Sunil V.
Sharma
a,
Cristina
Pubill-Ulldemolins
ab,
Joshua A.
Sharp
a,
Christopher
Cartmell
a,
Paco
Cárdenas
 c and
Rebecca J. M.
Goss
*a
c and
Rebecca J. M.
Goss
*a
aDepartment of Chemistry & BSRC University of St Andrews St Andrews, KY16 9ST, UK. E-mail: rjmg@st-andrews.ac.uk
bDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton, BN19QJ, UK
cDepartment of Medicinal Chemistry, Uppsala University, Uppsala 75123, Sweden
First published on 8th October 2019
Abstract
Blending synthetic biology and synthetic chemistry represents a powerful approach to diversity complex molecules. To further enable this, compatible synthetic tools are needed. We report the first Buchwald Hartwig amination reactions with unprotected halotryptophans under aqueous conditions and demonstrate this methodology is applicable also to the modification of unprotected tripeptides and the natural product barettin.
Tryptophan is an essential amino acid, abundant within peptides and proteins, and central to their fluorescence, folding and structure;1 it is also an important component in many bioactive natural products including non-ribosomal peptides e.g., vancomycin, and a precursor to indole alkaloids e.g., vinblastine and vincristine.2 Modulation of tryptophan affords the potential to tune fluorescence, conformation and activity of these (bio)molecules.3 Although there has been significant research exploring the cross-coupling chemistry of nucleosides and the aromatic amino acid phenylalanine, both in their free state or as components of larger systems, the cross-coupling of halotryptophan has seemingly received little attention. Methodology now exists for the modification of halotryptophans, by Sonogashira4 and Suzuki-Miyaura5,6 but, to the best of our knowledge, the utilisation of Buchwald Hartwig amination (BHA) of halotryptophans and their derivatives remains unexplored.
Whilst BHA conditions have been applied to a wide range of aromatic species including indoles and 6-bromoquinolone (Scheme 1A and B),7,8 to the best of our knowledge there is no literature precedent for its use in unprotected biomolecule functionalisation. Herein we report the application of aqueous BHA coupling to 5-bromoindole (Scheme 1C) and unprotected halotryptophans with a range of substituted aniline coupling partners (Scheme 1D). We then present the BHA derivatisation of tripeptides and the natural product barettin (Scheme 1E and F).
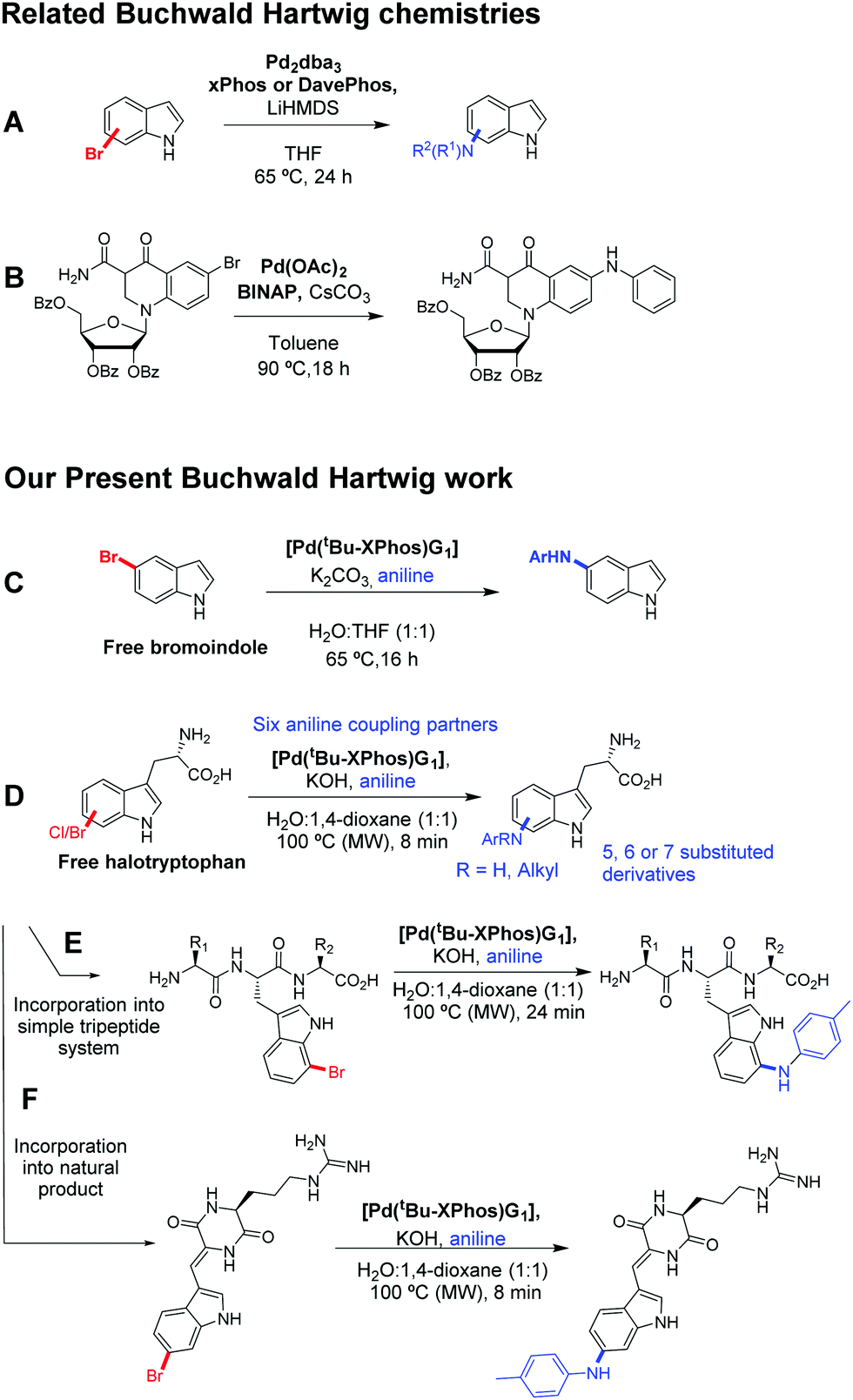 | ||
| Scheme 1 Buchwald Hartwig related work: examples of relevant prior work are outlined along with work within this study. (A) Example literature conditions for BHA on a bromoindole from Buchwald.7 (B) Buchwald Hartwig coupling for a 6-bromoquinolone nucleoside, Wicke et al.8 (C) Reported BHA modification for 5-bromoindole. (D) Reported BHA modifications for a range of halotryptophans. (E) Reported BHA modification of a bromotryptophan containing tripeptide. (F) Reported BHA modification the natural product barettin. | ||
We set out to explore the feasibility of developing aqueous conditions for BHA coupling of halotryptophans, which may be readily accessed through a simple one-step biotransformation of indoles using tryptophan synthase9 or through a 4–5 step chemical synthesis.10 One challenge presented by free amino acids in palladium coupling reactions is the amino acid's ability to chelate and deactivate the palladium catalyst.11 To reduce the likelihood of catalyst deactivation, we selected a precatalyst, L–Pd–G1 (2) (Fig. 1) with the tBu-XPhos ligand, as shown by Buchwald and co-workers.12 Initially, we examined the coupling of unprotected 5-bromoindole 3 and aniline 4.7 Investigating the impact of varying aqueous solvent mixture, catalyst loading, aniline eq. and base screening led us to conditions of 2 eq. of aniline, 2 mol% [Pd(tBu-XPhos)G1] catalyst, 1.2 eq. K2CO3, THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (1:1) at 65 °C for 16 h (Scheme 2) giving an 85% yield of 5.
:water (1:1) at 65 °C for 16 h (Scheme 2) giving an 85% yield of 5.
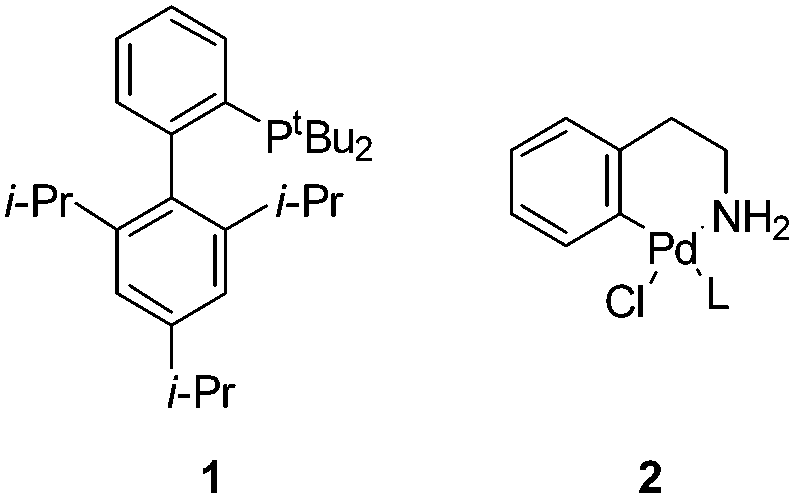 | ||
| Fig. 1 Buchwald ligand tBu-XPhos 1 and precatalyst L–Pd–G12. | ||
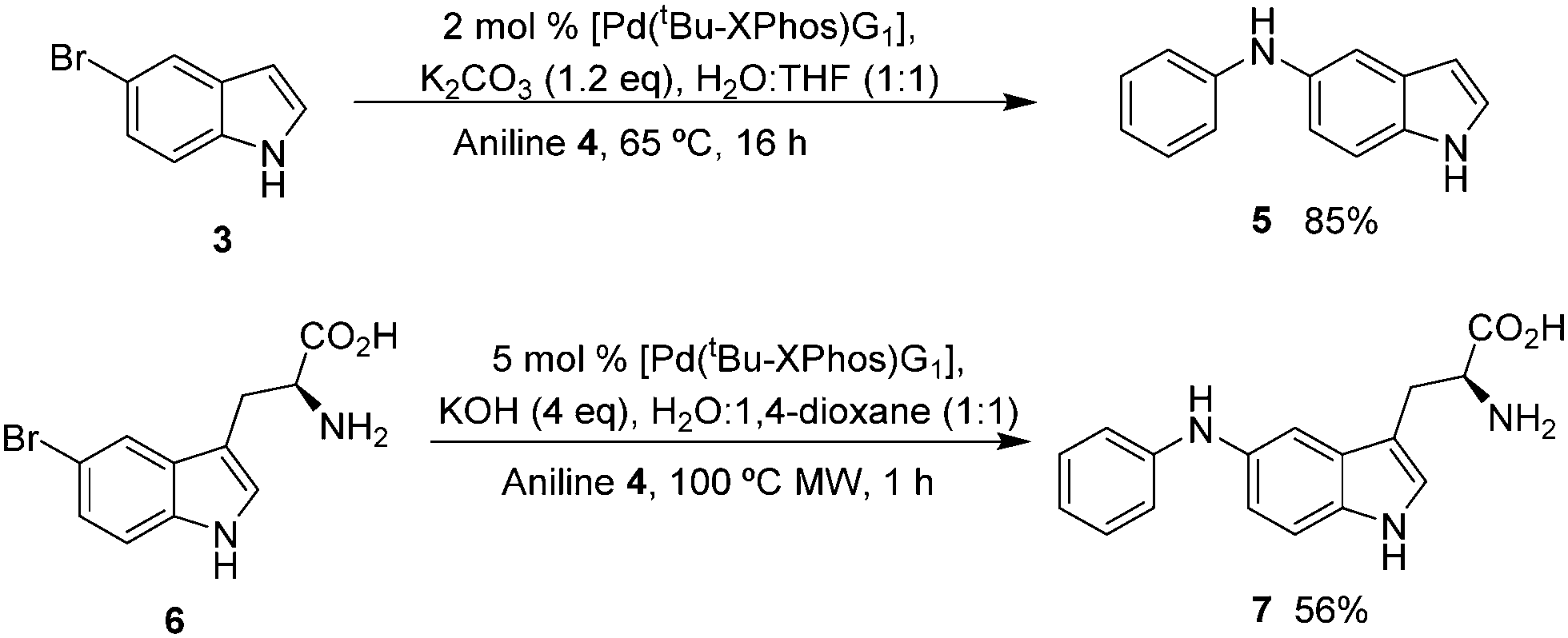 | ||
| Scheme 2 Buchwald Hartwig Amination reaction of 5-bromoindole (3) and 5-bromotryptophan (6) with aniline (4). | ||
Translating these conditions directly to the more challenging cross-coupling of 5-bromotryptophan, we saw a substantial drop in yield. This prompted a further round of optimisation and a move to microwave heating resulting in 56% isolated yield of the cross-coupled product 7 (Scheme 2).
Next, we decided to take a closer look at the influence of reaction time under the microwave conditions exploring the impact of heating by microwave from 1 minute to 1.5 h (Table 1).
| Time of reaction (min) | Yield (%) |
|---|---|
| Conditions: 5-bromotryptophan (1 eq.), [Pd(tBu-XPhos)G1] (5 mol%), KOH (4.0 eq.), aniline (2.0 eq.), H2O/1,4-dioxane (1:1, 0.6 mL), microwave heating, sealed tube, under argon, 100 °C.a Isolated yield of the product after reverse phase purification.b Yield assessed by 1H-NMR using 1,3,5-tri-tert-butylbenzene as an internal standard. |
|
| 1 | 43a |
| 5 | 53a |
| 8 | 75a |
| 15 | 71a |
| 30 | 71a |
| 60 | 56b |
| 90 | 35b |
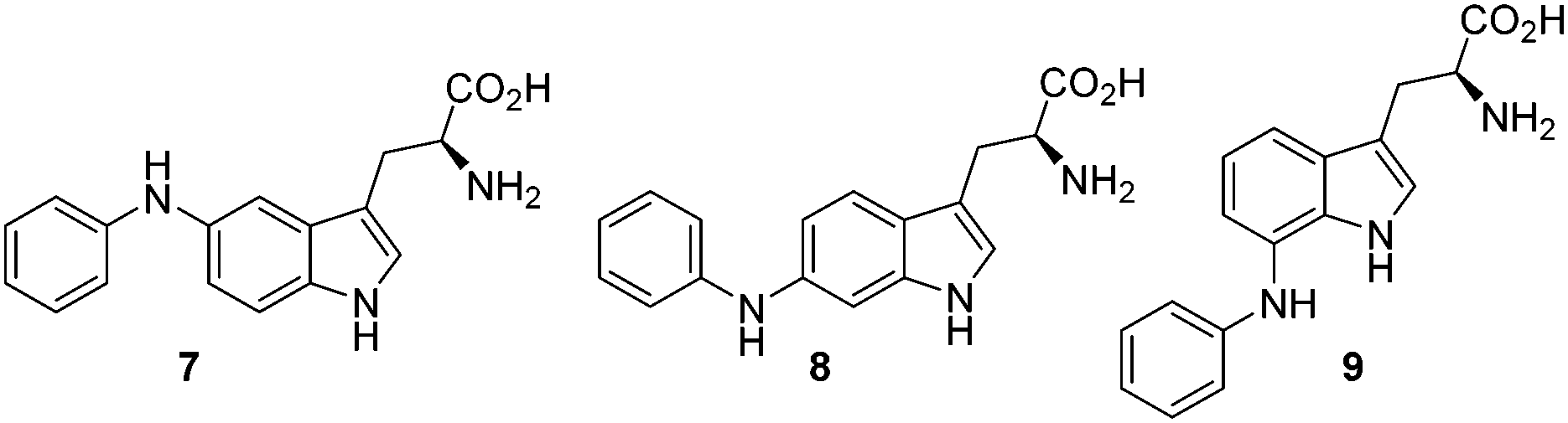
We found that shorter reaction times gave improved yields, optimum at 8 min (75% yield) (Table 1). It is suspected that longer reaction times cause degradation of the product resulting in lower yields, no clear by-products could be identified. Having established utilisable conditions for the BHA of 5-bromotryptophan, we investigated the reaction with tryptophans substituted with bromo and chloro in varying positions (Table 2). The reaction proceeded well for 5, 6 and 7-bromotryptophans resulting in yields of 75, 51 and 68% respectively. Similarly, the 5, 6 and 7-chlorotryptophans also worked well (67, 60 and 31% yields respectively).
|
|
|||
|---|---|---|---|
| Entry | Tryptophan | Product | Isolated yield/% |
| Conditions: halotryptophan (1 eq.), [Pd(tBu-XPhos)G1] (5 mol%), KOH (4.0 eq.), aniline (2.0 eq.), H2O/1,4-dioxane (1:1, 0.6 mL), microwave heating, sealed tube, under argon, 8 min, 100 °C. |
|||
| 1 | 5-Br | 7 | 75 |
| 2 | 6-Br | 8 | 51 |
| 3 | 7-Br | 9 | 68 |
| 4 | 5-Cl | 7 | 67 |
| 5 | 6-Cl | 8 | 60 |
| 6 | 7-Cl | 9 | 31 |
Having assessed reactivity of the various halotryptophans with aniline, we next sought to examine the effects of steric and electronic properties of the aniline coupling partner. We selected six anilines, 10 to 15, (Scheme 3 and Table 3) and reacted these with 5- and 7-bromotryptophan; the two most reactive regioisomers according to our positional scoping.
 | ||
| Scheme 3 Investigating the reaction of 5-bromo and 7-bromotryptophans with a selection of anilines. | ||
| Entry | Tryptophan | Product | Isolated yield/% |
|---|---|---|---|
| Conditions: bromotryptophan (1 eq.), [Pd(tBu-XPhos)G1] (5 mol%), KOH (4.0 eq.), anilines (2.0 eq.), H2O/1,4-dioxane (1:1, 0.6 mL), microwave heating, sealed tube, under argon, 8 min, 100 °C. |
|||
| 1 | 5-Br | 10a | 59 |
| 2 | 7-Br | 10b | 53 |
| 3 | 5-Br | 11a | 63 |
| 4 | 7-Br | 11b | 62 |
| 5 | 5-Br | 12a | No reaction |
| 6 | 7-Br | 12b | No reaction |
| 7 | 5-Br | 13a | No reaction |
| 8 | 7-Br | 13b | 4 |
| 9 | 5-Br | 14a | 7 |
| 10 | 7-Br | 14b | 15 |
| 11 | 5-Br | 15a | 67 |
| 12 | 7-Br | 15b | 59 |
These results show that cross-coupling proceeded even in the presence of moderate steric hindrance: 2-methylaniline (11) reacted with 5- and 7-bromotryptophans nearly as efficiently as unsubstituted aniline, with yields of 63% and 62% respectively. However, it is striking that two methyl groups in close proximity to the aryl amine, as in 2,6-dimethylaniline (12), led to complete suppression of the reaction and recovery of starting materials. Interestingly, N-methylaniline (13) is partially tolerated yielding 4% product for the 7-bromotryptophan, but no product could be obtained for the 5-bromotryptophan.
Differently substituted anilines, bearing electron-withdrawing or electron-donating groups, were subsequently tested to assess the impact of the electronic properties of the nucleophile in our reaction. With the weakly electron donating para-methyl substituent, we saw a slight drop in yield from 75% and 68% to 59% and 53% respectively for the 5- and 7-bromotryptophans. It was predicted13 that the strong electron withdrawing nature of the CF3 substituent would reduce the reactivity of the aniline towards amination and this was borne out in the very low isolated yields (7 and 15%, Table 3, entries 9 and 10). Also, as anticipated, the electron rich trimethoxyaniline reacted well (67 and 59% yields Table 3, entries 11 and 12), but did not surpass the yields observed for the unsubstituted anilines reactions. The slightly lower than expected yield for 15 may be due to improved organic solubility of the products resulting in a slight loss of product during the work up.
We next tested the reaction with more complex halotryptophan containing substrates. Tripeptides 16, 17 and 18 were synthesised (see ESI†) and were subjected to our BHA conditions (Scheme 4) using p-toluidine (10) as the coupling partner and increasing the reaction times to allow for potentially slower reaction with the more complex molecules. Tripeptide 17 reacted well and a 99% isolated yield was achieved. The products of tripeptides 16 and 18 suffered losses during purification. In particular peptide 21 had poor stability in solution and required careful handling to isolate high purity material. These examples demonstrate the successful BHA coupling of halotryptophan present at either terminal or middle locations in an unprotected peptide sequence possessing polar/non-polar side chains.
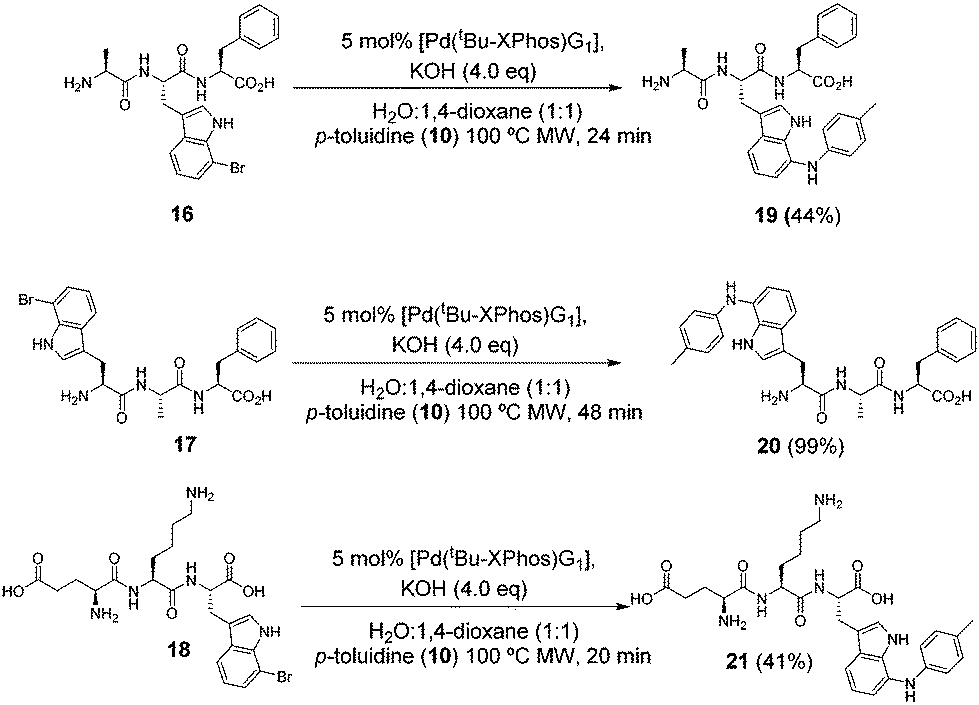 | ||
| Scheme 4 Investigating the reaction of 7-bromotryptophan containing tripeptides with p-toluidine (10). | ||
Encouraged by these results, we next chose derivatisation of barettin a marine natural product reputed to have antioxidant and anti-inflammatory properties and containing a 6-bromotryptophan group. Using our standard conditions (Scheme 5) we observed full consumption of starting material. LCMS showed a distinct peak at 4.3 min with an [M + H]+ of 446 corresponding to the desired product. Despite the small scale and purification difficulties, we were able to isolate 1 mg of the product 23 following HPLC purification, (purity >95%, 16% yield).
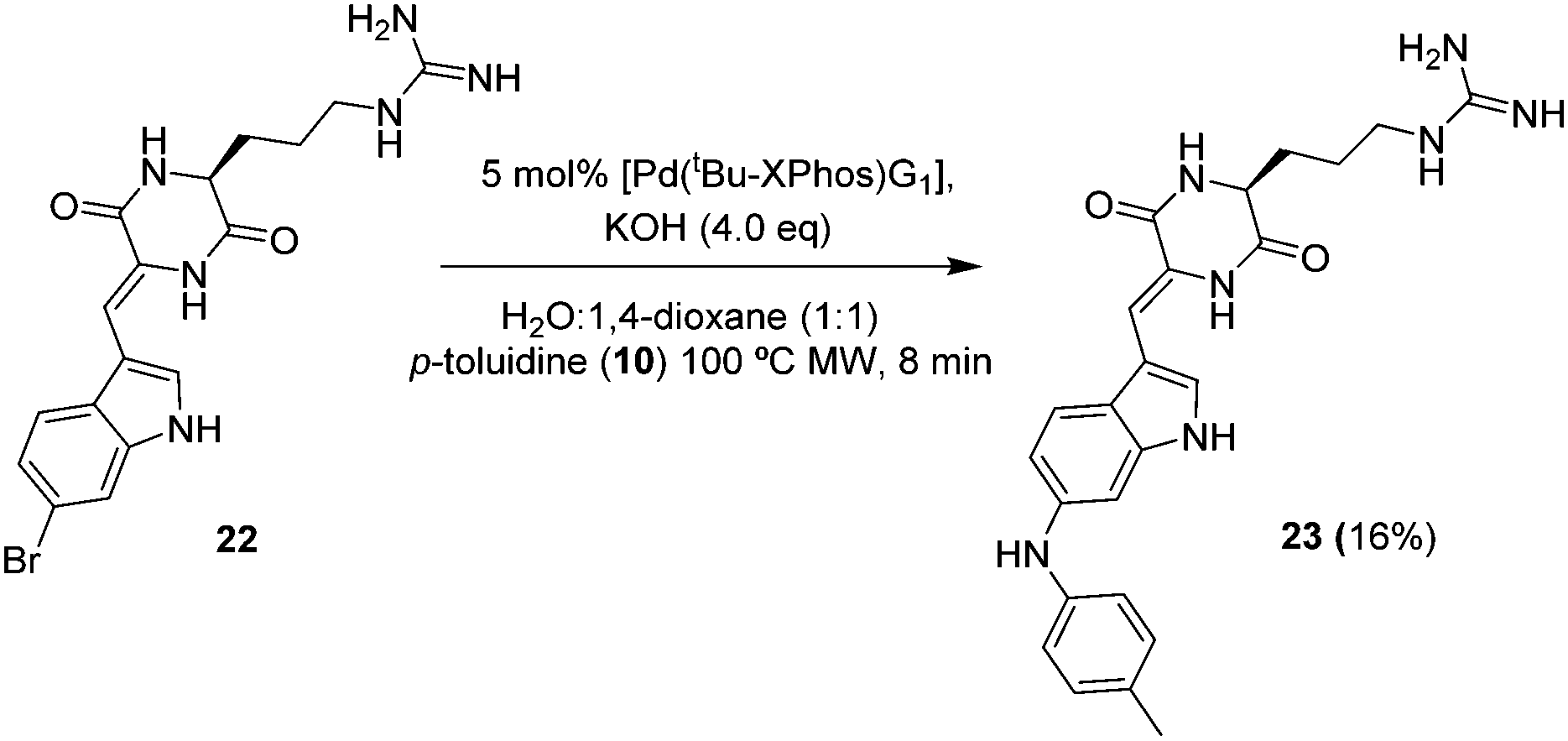 | ||
| Scheme 5 BHA reaction of barettin with p-toluidine (10). | ||
We have for the first time demonstrated the use of Buchwald Hartwig Amination to diversify free unprotected 5, 6 and 7-bromo- and chlorotryptophans, and shown the reaction is also effective on short 7-bromotryptophan containing peptides including potentially reactive groups such as glutamic acid and lysine. The reaction products can be isolated by reverse-phase chromatography or HPLC. As seen in our previous work,6b following the cross-coupling reaction, the tripeptides were single peaks by HPLC(LCMS) (see ESI†) indicating presence of single diastereomers and therefore no observable epimerisation occurred during the cross-coupling reactions. Our new conditions were also successfully applied to the more complex natural product barettin. This provides an expansion of the reactions that can be carried out with easily accessed9 halotryptophans that can be incorporated into more complex peptides and natural products. The incorporation of the halotryptophans and wide range of amine coupling partners could be envisioned to produce a wide variety of natural product analogues for use in structure–activity relationship analyses.
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013/ERC grant agreement no. 614779 GenoChemetics (to R. J. M. G.)). P. Cárdenas received support from the European Union's Horizon 2020 research and innovation program through the SponGES project (grant agreement no. 679849). This document reflects only the authors’ view and the Executive Agency for Small and Medium-sized Enterprises (EASME) is not responsible for any use that may be made of the information it contains. We thank the National Mass Spectrometry service at Swansea for MS characterisation of synthetic products.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) G. D. Rose, A. R. Geselowitz, G. J. Lesser, R. H. Lee and M. H. Zehfus, Science, 1985, 229, 834–838 CrossRef CAS PubMed; (b) D. A. Dougherty, Science, 1996, 271, 163–168 CrossRef CAS PubMed.
- R. L. Noble, C. T. Beer and J. H. Cutts, Ann. N. Y. Acad. Sci., 1958, 76, 882–894 CrossRef CAS PubMed.
- (a) R. J. M. Goss, S. Shankar and A. A. Fayad, Nat. Prod. Rep., 2012, 29, 870–889 RSC; (b) K. P. P. Mahoney, D. R. M. Smith, E. J. A. Bogosyan and R. J. M. Goss, Synthesis, 2014, 2122–2132 CAS; (c) A. E. Osbourn, R. J. M. Goss and G. T. Carter, Natural Products: Discourse, Diversity, and Design, Wiley-Blackwell, 2014, ISBN: 978-1-118-29806-0 CrossRef; (d) A. Kirschning, F. Taft and T. Knobloch, Org. Biomol. Chem., 2007, 5, 3245–3259 RSC.
- M. J. Corr, S. V. Sharma, C. Pubill-Ulldemolins, R. T. Bown, P. Poirot, D. R. M. Smith, C. Cartmell, A. Abou Fayad and R. J. M. Goss, Chem. Sci., 2017, 8, 2039–2046 RSC.
- A. Deb Roy, R. J. M. Goss, G. K. Wagner and M. Winn, Chem. Commun., 2008, 4831–4833 Search PubMed.
- (a) A. Deb Roy, S. Grüschow, N. Cairns and R. J. M. Goss, J. Am. Chem. Soc., 2010, 132, 12243–12245 CrossRef PubMed; (b) S. V. Sharma, X. Tong, C. Pubill-Ulldemolins, C. Cartmell, E. J. A. Bogosyan, E. J. Rackham, E. Marelli, R. B. Hamed and R. J. M. Goss, Nat. Commun., 2017, 8, 229–238 CrossRef PubMed.
- M. D. Charles, P. Schultz and S. L. Buchwald, Org. Lett., 2005, 7, 3965–3968 CrossRef CAS PubMed.
- L. Wicke, J. W. Engels, R. Gambari and A. M. Saab, Arch. Pharm. Chem. Life Sci., 2013, 346, 757–765 CrossRef CAS PubMed.
- (a) D. R. M. Smith, T. Willemse, D. S. Gkotsi, W. Schepens, B. U. W. Maes, S. Ballet and R. J. M. Goss, Org. Lett., 2014, 16, 2622–2625 CrossRef CAS PubMed; (b) R. J. M. Goss and P. L. A. Newill, Chem. Commun., 2006, 4924–4925 RSC.
- For example, see: (a) Y. Konda-Yamada, C. Okada, K. Yoshida, U. Yasuyuki, S. Arima, N. Sato, T. Kai, H. Takayanagi and Y. Harigaya, Tetrahedron, 2002, 58, 7851–7861 CrossRef CAS; (b) C. Ma, X. Liu, X. Li, J. Flippen-Anderson, S. Yu and J. Cook, J. Org. Chem., 2001, 66, 4525–4542 CrossRef CAS PubMed.
- M. A. Carvalho, B. C. Souza, R. E. F. Paiva, F. R. G. Bergamini, A. F. Gomes, F. C. Gozzo, W. R. Lustri, A. L. B. Formiga, G. Rigatto and P. P. Corbi, J. Coord. Chem., 2012, 65, 1700–1711 CrossRef CAS.
- N. C. Bruno, M. T. Tudge and S. L. Buchwald, Chem. Sci., 2013, 4, 916–920 RSC.
- K. H. Hoi, S. Calimsiz, R. D. J. Froese, A. C. Hopkinson and M. G. Organ, Chem. – Eur. J., 2012, 18, 145–151 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures; NMR characterisation; LC-MS characterisation. See DOI: 10.1039/c9cc02554e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |