
Direct biocatalysed synthesis of first sulfur-, selenium- and tellurium- containing L-ascorbyl hybrid derivatives with radical trapping and GPx-like properties†
Damiano
Tanini
 *,
Beatrice
Lupori
,
Gianni
Malevolti
,
Moira
Ambrosi
,
Pierandrea Lo
Nostro
and
Antonella
Capperucci
*
*,
Beatrice
Lupori
,
Gianni
Malevolti
,
Moira
Ambrosi
,
Pierandrea Lo
Nostro
and
Antonella
Capperucci
*
University of Florence, Department of Chemistry “Ugo Schiff”, Via della Lastruccia 13, I-50019 Sesto Fiorentino, Italy. E-mail: damiano.tanini@unifi.it; antonella.capperucci@unifi.it
First published on 17th April 2019
Abstract
6-O-L-Ascorbyl selenoesters, thioesters and telluroesters can be efficiently and directly prepared from L-ascorbic acid and suitable functionalised chalcogenoesters through lipase-catalysed transesterification reactions. Novel synthesised L-ascorbyl derivatives exhibited remarkable chain breaking and glutathione peroxidase-like activities.
L-Ascorbic acid (vitamin C) is a powerful water soluble antioxidant essential for the appropriate functioning of the body, and is involved in a number of biological processes ranging, amongst others, from the biosynthesis of collagen1 and catecholamine to the modulation of neurotransmission.2
The enhanced concentration of harmful reactive oxygen species (ROS) has long been related to the onset of several human diseases such as cancer, immune disorders, cystic fibrosis, and neurodegenerative diseases.3 Liposoluble vitamin E and hydrosoluble vitamin C, together with phenolic compounds, carotenoids, and trace elements such as zinc and selenium, are the main exogenous defences against oxidative stress. In this context, the design and development of novel antioxidants have been attracting growing interest over the past few decades.4 Particularly, owing to their capability to mimic the glutathione peroxidase activity, the synthesis of chalcogen-containing antioxidants has recently attracted growing attention. Furthermore, organochalcogenides can possess anticancer, antibacterial, and enzyme inhibition activities.5 The functionalization of bioactive natural products with chalcogens represents an effective strategy to modulate or improve their biological properties. For example, the introduction of chalcogen-containing moieties into natural compounds such as tocopherols,6,7 tocotrienols,7 retinol,8 hydroxytyrosol,9 chrysin,10 quercetin,10a and resveratrol10c has been extensively studied. However, whilst sulfur-, selenium-, or tellurium-functionalised vitamin E and vitamin A derivatives have been reported, to the best of our knowledge, the synthesis of chalcogen-containing vitamin C derivatives has never been described. The paucity of such results is reasonably due to the instability of the L-ascorbic acid core, which easily undergoes oxidation of the enediol moiety and ring opening of the lactone. Furthermore, several alcohol protecting group strategies cannot be applied since the conditions of the endgame protecting group cleavage are often not compatible with chalcogens or with the L-ascorbic acid core. For example, because the presence of chalcogens poisons the Pd-catalyst,11 the most commonly employed benzyl protecting group cannot be cleaved under mild Pd/C catalysed hydrogenation conditions. Additionally, harsh bases- or acid-mediated deprotection procedures cause severe decomposition of products.
We sought to approach this problem from a different perspective and evaluated the possibility of applying lipase biocatalysed transesterifications12 of chalcogen-containing esters and L-ascorbic acid. This approach would ideally allow a straightforward access to chalcogen-containing L-ascorbic acid derivatives, without requiring tedious and detrimental protection/deprotection steps. Lipase B catalysed transesterification is indeed a versatile tool to synthesise L-ascorbyl esters under green and mild conditions.13 However, to the best of our knowledge, its application to chalcogen-containing acyl donors has never been described and its feasibility and functional group tolerance were not obvious.
We commenced our studies by establishing the optimal conditions required to promote the biocatalysed transesterification reaction of β-selenoester 2a, prepared through seleno-Michael addition from benzeneselenol 1a and methyl acrylate (see ESI†), with L-ascorbic acid 3. According to a literature survey, the optimal temperature for the enzyme activity was established to be 45 °C. Polar solvents, such as acetone and tertiary alcohols, are the media of choice for lipase-catalysed L-ascorbic acid esterifications.12,13 In our work, acetone proved to be the most effective, plausibly owing to its high capability to dissolve both 2a and 3. In addition non protic solvents such as acetone do not promote the dissociation of L-ascorbic acid to the more oxidisable L-ascorbate. Evaluation of different solvents commonly used for lipase B catalysed reactions, such as tert-butanol and 2-methyl-1-butanol, gave lower yields. Thus, we investigated the effect of the reaction stoichiometry and the amount of lipase. We found that, whilst poor yields were obtained upon using 500 U mmol−1 of enzyme (Table 1, entries 1–3), doubling of the enzyme amount brought about a significant improvement in the yield (Table 1, entry 4).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | 2a (eq.) | Lipase B (U mmol−1) | Time (h) | Yielda (%) | Entry | 2a (eq.) | Lipase B (U mmol−1) | Time (h) | Yielda (%) |
| a Isolated yield is reported. b Partial decomposition of 4a was observed. c 4 A molecular sieves (300 mg) were added. See ESI for details. The use of molecular sieves with a lower enzyme loading (500 U mmol−1) resulted in a slight improvement in yield. | |||||||||
| 1 | 1.0 | 500 | 48 | 12 | 6 | 0.3 | 1000 | 24 | <10 |
| 2 | 0.5 | 500 | 48 | <10 | 7 | 1.0 | 1000 | 48 | 26 |
| 3 | 0.3 | 500 | 48 | <10 | 8 | 3.0 | 1000 | 48 | 45 |
| 4 | 0.3 | 1000 | 48 | 22 | 9 | 3.0 | 1000 | 48 | 74c |
| 5 | 0.3 | 1000 | 72 | 14b | 10 | 0.3 | 1000 | 48 | 32c |
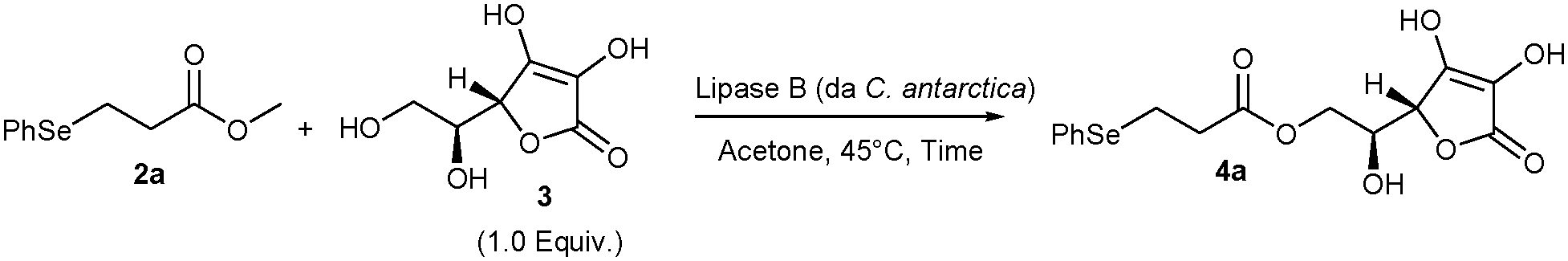
The optimal reaction time was found to be 48 h, as partial decomposition of the reaction product was observed after 72 h (Table 1, entry 5). On the other hand, shorter times resulted in much lower yields (Table 1, entry 6). Pleasingly, we found that by using an excess amount of β-selenoester 2a the desired 6-O-L-ascorbyl ester 4a was formed in a rather good yield for this type of biocatalysed transformation (45%, Table 1, entry 8).
Remarkably, a significant improvement in yield was achieved upon performing the reaction in the presence of 4 Å molecular sieves (Table 1, entries 9 and 10). Particularly, by using an excess amount of 2a under these conditions, the ascorbyl derivative 4a was formed in 74% yield, which represents a very good result for lipase-catalysed transesterification reactions. The striking effect of zeolites can be reasonably ascribed to the trapping of both MeOH and H2O. Indeed, the removal of methanol produced by the transesterification displaces the equilibrium of the reaction toward the formation of 4a. Furthermore, by trapping water, molecular sieves hamper the competitive lipase-catalysed hydrolysis of 4a.
Having identified the optimal reaction conditions, we proceeded to investigate the scope of the transformation with respect to different chalcogen-containing esters. Thus, a large variety of differently substituted selenium-, sulfur-, and tellurium-containing esters (Scheme 1 and Scheme S1, ESI†) were synthesised as shown in Scheme 1. β-Arylseleno- and β-alkylseleno-esters 2a–g were smoothly prepared through a novel seleno-Michael addition14 involving suitable aryl- or alkyl-selenols151 and methyl acrylate. The reaction occurred under very mild conditions in the presence of Al2O3 (Scheme 1 and Scheme S1 (ESI†), via A). Furthermore, α-selenoesters 2h and 2i and γ-selenoester 2j were easily obtained by exploiting the reactivity of selenols 1 with methyl bromoacetate and ethyl 4-bromobutyrate, respectively (Scheme 1 and Scheme S1 (ESI†), via B). Similarly, variously substituted and functionalised α-, β-, and γ-thioesters 6a–i were prepared from the corresponding aryl or alkyl thiols16a and suitable electrophiles (Scheme S1 (ESI†), via A and via B). Disulfide 6j, bearing two ester functions and four sulfur atoms, was synthesised from 1,9-nonanethiol through a two-step procedure involving thiol-Michael addition and a DCF (dicyanofumarate) mediated oxidation sequence (see ESI† for details).16b β-Aryltelluroesters 8a,b were prepared from the corresponding ditellurides and methyl 3-bromopropionate (Scheme 1 and Scheme S1, ESI†).
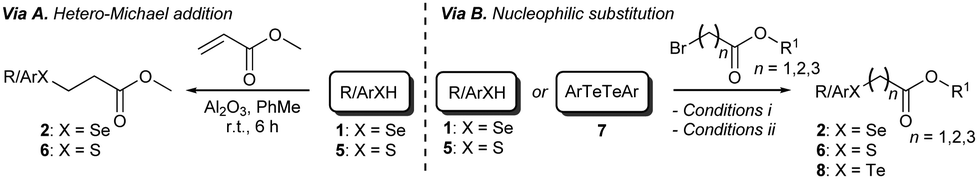 | ||
| Scheme 1 Synthesis of selenoesters 2, thioesters 6, and telluroesters 8. See ESI† for reagents and conditions, and experimental details. | ||
With a range of differently substituted and functionalised chalcogen-containing esters in hand, we then explored the scope of the lipase-catalysed transesterification reaction with L-ascorbic acid (Scheme 2). Under the optimised reaction conditions, o-, m-, and p-methoxy phenylselenoesters 2b–d gave selectively the corresponding 6-O-ascorbyl arylselenoalkanoates 4b–d in good yields. β-Selenoester 2e, bearing a p-F-C6H4 ring, also reacted efficiently with 3 to afford compound 4e. The reaction was also successfully applied to the synthesis of L-ascorbyl derivatives 4f and 4g bearing different alkylseleno moieties, including the functionalised glycidol derivative (4g). Furthermore, methyl and ethyl α- and γ-selenoesters 2h–j were efficiently transesterified with 3 yielding the corresponding 6-O-L-ascorbyl esters 4h–j in rather good yields, therefore demonstrating the versatility of the biocatalysed approach towards the synthesis of variously functionalised homologous L-ascorbyl selenoesters. Next, we turned our attention to evaluating the generality of such a reaction with respect to sulfur-containing esters. β-Arylthioesters 6a–d were smoothly converted into the corresponding sulfurated 6-O-ascorbyl derivatives bearing a phenyl ring (9a) or o- (9b), m- (9c), and p- (9d) bromo-substituted benzenes bonded to the S atom. This methodology could also be applied to more interesting highly functionalised alkyl sulfides. The hydroxy-substituted S-alkyl β-thioester 6e and the enantio-enriched epichlorohydrin derivatives (S)-6f and (R)-6f were successfully transferred onto the L-ascorbic acid core, affording the corresponding functionalised ester 9e and the enantio-enriched derivatives 9f and 9g, containing three controlled stereogenic centers and a further functionalisable chlorinated chain. Additionally, a chiral enantio-enriched N-tosyl amino-substituted β-thioester 6g, synthesised from L-valine,14,15a could be efficiently employed to access the enantio-enriched S,N-containing 6-O-L-ascorbyl ester 9h. Furthermore, α- and γ-thioesters 9i and 9j were also conveniently obtained by treating 3 with 6h and 6i, respectively, under the above described conditions. Remarkably, the disulfide 6j was successfully used in a double biocatalysed transesterification to afford the potentially valuable bola type bis-ascorbyl ester 9k, bearing two L-ascorbyl moieties and four sulfur atoms. Indeed, owing to the unique physicochemical properties of bola type structures, novel bifunctional chalcogen-containing amphiphilic ascorbic acid derivatives could find wide applications both in polymers and materials science and in medicinal chemistry for the drug delivery of lipophilic molecules.12a,17
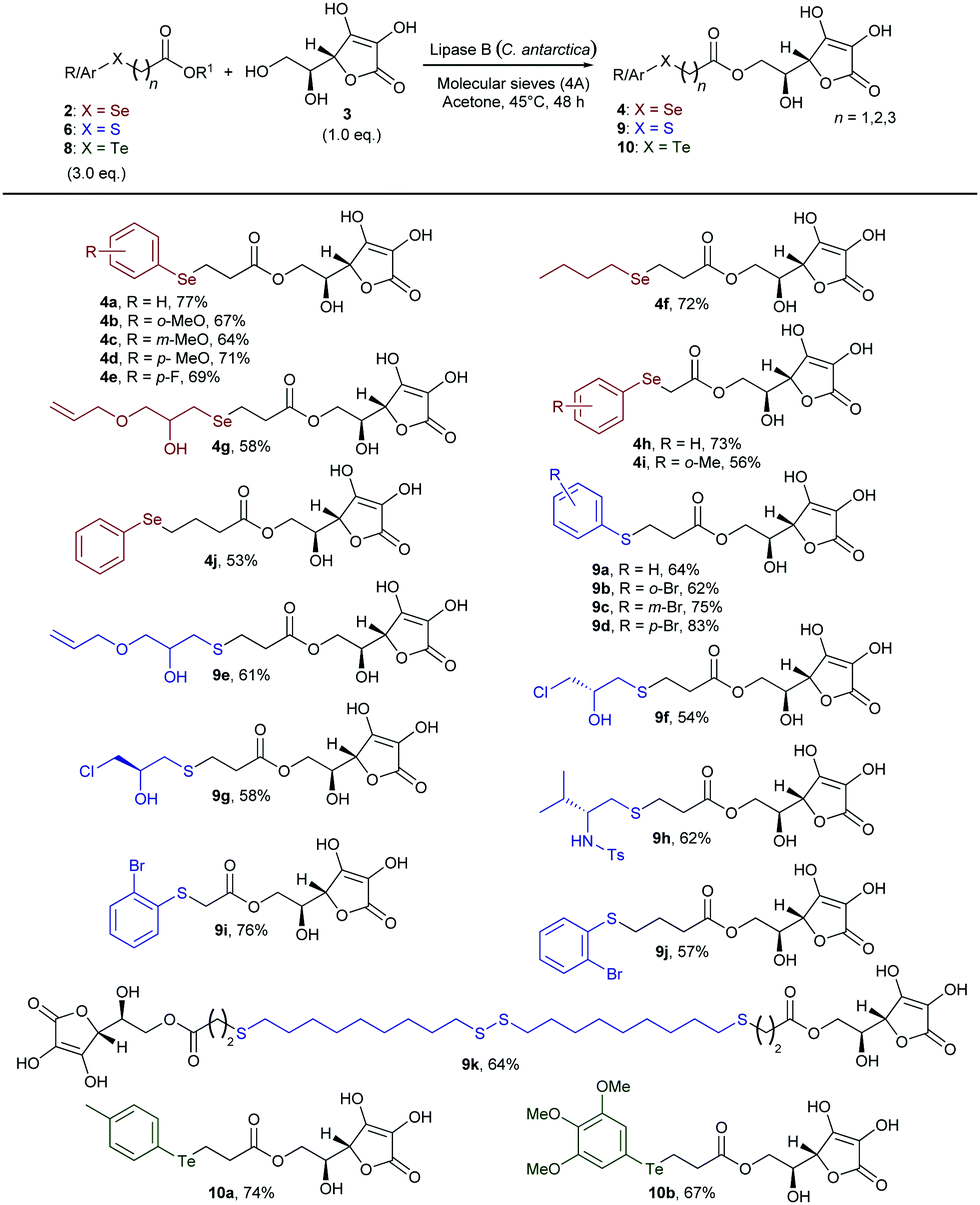 | ||
| Scheme 2 Biocatalysed direct synthesis of selenium-, sulfur-, and tellurium-containing 6-O-ascorbyl esters 4, 9, and 10 from L-ascorbic acid. Isolated yields are reported. | ||
Finally, in order to extend the scope of this methodology to organotellurium derivatives, we evaluated the reactivity of β-aryltelluroesters 8a and 8b with 3. 6-O-L-Ascorbyl aryltelluroalkanoates 10a and 10b were directly and selectively formed in good yield, thus highlighting the remarkably broad scope of the procedure and the possibility to use the lipase-catalysed approach with all chalcogens.
Owing to the presence of both the vitamin C free enediol moiety and the selenium or tellurium atom, ascorbyl derivatives 4 and 10 can exhibit both chain breaking and catalytic antioxidant activities, therefore representing excellent antioxidant candidates. Therefore, having developed a convenient procedure to access novel chalcogen-containing L-ascorbic acid derivatives, we wished to investigate their antioxidant properties. Pleasingly, according to the DPPH assay, all the synthesised 6-O-ascorbyl esters exhibited remarkable chain breaking activity,18 leading to a rapid free radical quenching (complete decolouration of ethanolic DPPH˙ solution occurred within 3 seconds) and, thus, showing radical scavenger properties comparable to those of L-ascorbic acid, which represents the most powerful hydrophilic antioxidant. The stoichiometry of the reaction (n, number of radical trapped within 10 minutes) was found to be n = 2 for all the tested L-ascorbyl esters 4, 9, and 10, thus indicating that under the experimental conditions the oxidation products of these compounds are not capable of reacting with DPPH. Furthermore, the catalytic thiol peroxidase like activity of selected L-ascorbyl derivatives was also pursued according to methods reported in the literature using dithiothreitol (DTT)19 or glutathione (GSH)19 as substrates (see Scheme S2, and Fig. S2 and S3, ESI†). We were delighted to discover that all tested selenium- and tellurium-contained vitamin C derivatives behave as catalysts in promoting the reduction of hydrogen peroxide in the presence of a thiol cofactor (Table 2). Intriguingly, when the DTT oxidation assay is accomplished using 1.0 eq. of hydrogen peroxide ca. 10–20% of DTT remained unreacted. Control experiments suggested that reaction of the L-ascorbate moiety with H2O2 proceeds slowly in the absence of catalysts. The observed results might be reasonably ascribed to the formation of tricarbonyl derivatives, proceeding through a redox reaction involving the enediol moiety and a selenoxide or telluroxide intermediate.20 On the other hand, complete DTT oxidation was achieved upon using an excess amount (1.2 eq.) of H2O2. As can be noticed (Table 2 and Fig. 1), under these conditions all the tested compounds exhibited catalytic thiol-peroxidase like properties, being able to promote the oxidation of both DTT and GSH. Intriguingly, the β-alkylselenoester 4g showed higher catalytic activity with respect to the arylseleno-substituted analogues 4a−c and 4e. Furthermore, according to both assays, L-ascorbyl β-aryltelluroalkanoates 10a and 10b behaved as more effective catalysts with respect to similar selenium-containing derivatives 4a–e.
| Entry | Compound | DTT (T50)ab | GSH/GR (T50)ac |
|---|---|---|---|
| a T 50 is the time required, in seconds, to halve the initial thiol concentration after the addition of H2O2; data in parentheses are the experimental error. b DTT oxidation was monitored by 1H NMR spectroscopy; 10 mol% of 4 and 1 mol% of 10 were used. c NADPH consumption was monitored by UV spectroscopy (340 nm). | |||
| 1 | 4a | 3405 (± 268) | 43 (± 5) |
| 2 | 4b | 4270 (± 325) | 48 (± 7) |
| 3 | 4c | 4046 (± 314) | 45 (± 5) |
| 4 | 4e | 3862 (± 362) | 53 (± 4) |
| 5 | 4g | 1346 (± 151) | 37 (± 3) |
| 6 | 4h | 2846 (± 116) | 42 (± 6) |
| 7 | 10a | 654 (± 104) | 18 (± 3) |
| 8 | 10b | 386 (± 93) | 14 (± 3) |
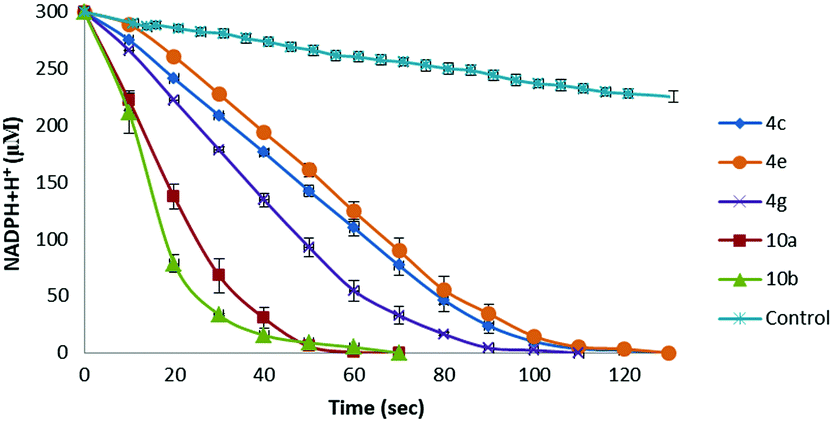 | ||
| Fig. 1 NADPH-coupled GPx assay. Reaction conditions: [NADPH]0 = 0.3 mM, [GSH]0 = 1.0 mM, [H2O2]0 = 2.5 mM, [GR] = 4 units per mL, [catalyst] = 0.1 mM in pH 7.4 phosphate buffer at ambient temperature. The mean ± SD values of three separate experiments are reported. | ||
Furthermore, particularly interesting are the results obtained through the GSH/GR/NADPH coupled test, which better reproduces the cellular environment (Fig. 1). Indeed, under these conditions the novel synthesised L-ascorbyl derivatives exhibited GPx-like activity, thus demonstrating the effective enhanced antioxidant properties of these novel amphiphilic systems, thus offering new opportunities for their potentially wide application in chemistry, biology, and materials science.
Preliminary physicochemical characterisation of these unreported amphiphilic antioxidants showed that most of them behave as semicrystalline solids. The evolution of the crystal and of the amorphous fraction depends on the thermal history of the sample, a common behaviour of polymers. As an example, the DSC curves obtained for 9c are shown in Fig. S1 (see ESI†). The crystalline fraction in 9c melted at about 75 °C producing an endothermic peak with ΔH = 21.6 J g−1. When the melt was slowly cooled to −20 °C (at 5 °C min−1), crystallisation occurred producing an exothermic peak at about 35.5 °C with ΔH = 28.2 J g−1. The crystalline fraction was found to melt between 40 °C and 100 °C upon heating. Instead, when the sample was cooled quickly down to −20 °C (at 50 °C min−1), no crystallisation was seen, and a cold crystallisation exothermic peak was detected in the following heating cycle followed by a broad endothermic melting peak. Remarkably, while this behaviour is typical for polymers, it is rather unusual for such small molecules and may represent an opportunity for their application in materials science.
In summary, we have found a convenient, green, mild, and direct route to synthesise novel chalcogen-containing L-ascorbyl derivatives, which would not have been accessible through classic methodologies. Owing to their enhanced chain breaking and catalytic antioxidant properties, these conjugate molecules represent potential valuable systems in biology, medicinal chemistry, and materials science.
We thank MIUR-Italy (“Progetto Dipartimenti di Eccellenza 2018–2022” allocated to Department of Chemistry “Ugo Schiff”).
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. S. Pinnell, Yale J. Biol. Med., 1985, 58, 553 Search PubMed.
- R. Figueroa-Méndez and S. Rivas-Arancibia, Front. Physiol., 2015, 6, 397 CrossRef PubMed.
- (a) J. K. Andersen, Nat. Rev. Neurosci., 2004, 5, S18 CrossRef PubMed; (b) R. C. S. Seet, C.-Y. J. Lee, E. C. H. Lima, J. J. H. Tan, A. M. L. Quek, W.-L. Chong, W.-F. Looi, S.-H. Huang, H. Wang, Y.-H. Chand and B. Halliwell, Free Radical Biol. Med., 2010, 48, 560 CrossRef CAS PubMed.
- (a) S. Yoshida, F. Kumakura, I. Komatsu, K. Arai, Y. Onuma, H. Hojo, B. G. Singh, K. I. Priyadarsini and M. Iwaoka, Angew. Chem., Int. Ed., 2011, 50, 2125 CrossRef CAS PubMed; (b) C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255 CrossRef CAS PubMed.
- (a) A. R. Patra, S. S. Roy, A. Basu, A. Bhuniya, A. Bhattachariee, S. Hajra, U. H. Sk, R. Baral and S. Bhattacharya, Sci. Rep., 2018, 8, 2194 CrossRef PubMed; (b) A. Angeli, D. Tanini, A. Capperucci and C. T. Supuran, Bioorg. Chem., 2018, 76, 268 CrossRef CAS PubMed; (c) A. Angeli, D. Tanini, A. Capperucci, G. Malevolti, F. Turco, M. Ferraroni and C. T. Supuran, Bioorg. Chem., 2018, 81, 642 CrossRef CAS PubMed; (d) A. Angeli, D. Tanini, A. Capperucci and C. T. Supuran, ACS Med. Chem. Lett., 2017, 8, 1213 CrossRef CAS PubMed.
- V. P. Singh, J. F. Poon and L. Engman, Org. Lett., 2013, 15, 6274 CrossRef CAS PubMed.
- P. S. Vraka, C. Drouza, M. P. Rikkou, A. D. Odysseos and A. D. Keramidas, Bioorg. Med. Chem., 2006, 14, 2684 CrossRef CAS PubMed.
- S. C. Welch and M. Gruber, J. Med. Chem., 1979, 22, 1532 CrossRef CAS PubMed.
- G. Rodríguez-Gutiérrez, F. Rubio-Senent, A. Gómez-Carretero, I. Maya, J. Fernández-Bolaños, G. G. Duthie and B. de Roos, Eur. J. Nutr., 2018 DOI:10.1007/s00394-018-1733-y.
- (a) S. F. Fonseca, D. B. Lima, D. Alves, R. G. Jacob, G. Perin, E. J. Lenardão and L. Savegnago, New J. Chem., 2015, 39, 3043 RSC; (b) I. L. Martins, C. Charneira, V. Gandin, J. L. Ferreira da Silva, G. C. Justino, J. P. Telo, A. J. S. C. Vieira, C. Marzano and A. M. M. Antunes, J. Med. Chem., 2015, 58, 4250 CrossRef CAS PubMed; (c) D. Tanini, L. Panzella, R. Amorati, A. Capperucci, E. Pizzo, A. Napolitano, S. Menichetti and M. d’Ischia, Org. Biomol. Chem., 2015, 13, 5757 RSC.
- P. Albers, J. Pietsch and S. F. Parker, J. Mol. Catal. A: Chem., 2001, 173, 275 CrossRef CAS.
- (a) C. Dolle, P. Magrone, S. Riva, M. Ambrosi, E. Fratini, N. Peruzzi and P. Lo Nostro, J. Phys. Chem. B, 2011, 115, 11638 CrossRef CAS PubMed; (b) G. Carrea and S. Riva, Angew. Chem., Int. Ed., 2000, 39, 222 CrossRef.
- (a) K. Nott, A. Brognaux, G. Richard, P. Laurent, A. Favrelle, C. Jérôme, C. Blecker, J. P. Wathelet, M. Paquot and M. Deleu, Prep. Biochem. Biotechnol., 2012, 42, 348 CrossRef CAS PubMed; (b) S. Park, F. Viklund, K. Hulta and R. J. Kazlauskas, Green Chem., 2003, 5, 715 RSC.
- D. Tanini, S. Scarpelli, E. Ermini and A. Capperucci, Adv. Synth. Catal., 2019 DOI:10.1002/adsc.201900168.
- (a) D. Tanini, C. Tiberi, C. Gellini, P. R. Salvi and A. Capperucci, Adv. Synth. Catal., 2018, 360, 3367 CrossRef CAS; (b) A. Angeli, D. Tanini, A. Nocentini, A. Capperucci, M. Ferraroni, P. Gratteri and C. T. Supuran, Chem. Commun., 2019, 55, 648 RSC.
- (a) D. Tanini, C. Borgogni and A. Capperucci, New J. Chem., 2019, 43, 6388 RSC; (b) G. Mlostoń, A. Capperucci, D. Tanini, R. Hamera-Fałdyga and H. Heimgartner, Eur. J. Org. Chem., 2017, 6831 CrossRef.
- S. Palma, R. H. Manzo, P. Lo Nostro and D. Allemandi, Int. J. Pharm., 2007, 345, 26 CrossRef CAS PubMed.
- (a) E. Tempestini, M. Bucci, V. Mastromartino, M. Gori, D. Tanini, M. Ambrosi, E. Fratini, A. Capperucci and P. Lo Nostro, ChemPhysChem, 2017, 18, 1400 CrossRef CAS PubMed; (b) P. Lo Nostro, G. Capuzzi, A. Romani and N. Mulinacci, Langmuir, 2000, 16, 1744 CrossRef CAS.
- (a) D. Tanini, A. Grechi, L. Ricci, S. Dei, E. Teodori and A. Capperucci, New J. Chem., 2018, 42, 6077 RSC; (b) F. Kumakura, B. Mishra, K. I. Priyadarsini and M. Iwaoka, Eur. J. Org. Chem., 2010, 440 CrossRef CAS.
- A reasonable catalytic cycle is the following. Ring opening of the oxidised lactone ring cannot be ruled out
.

Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and products’ characterisation. See DOI: 10.1039/c9cc02427a |
| This journal is © The Royal Society of Chemistry 2019 |