
Enantioselective synthesis of multi-nitrogen-containing heterocycles using azoalkenes as key intermediates
Liang
Wei
a,
Chong
Shen
a,
Yuan-Zhen
Hu
a,
Hai-Yan
Tao
a and
Chun-Jiang
Wang
 *ab
*ab
aEngineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China. E-mail: cjwang@whu.edu.cn; Fax: +86-27-68754067
bState Key Laboratory of Elemento-organic Chemistry, Nankai University, Tianjin 300071, China
First published on 16th May 2019
Abstract
Chiral multi-nitrogen-containing heterocycles, such as pyrazole, imidazole and pyridazine, are widely found in naturally occurring organic compounds and pharmaceuticals, and hence, their stereoselective and efficient synthesis is an important issue in organic synthesis. Out of the variety of methods that have been developed over the past century, the catalytic asymmetric cyclization and cycloaddition reactions are recognized as the most synthetically useful strategies due to their step-, atom- and redox-economic nature. In particular, the recently developed annulation reactions using azoalkenes as key intermediates show their great ability to construct diverse types of multi-nitrogen-containing heterocycles. In this feature article, we critically analyse the strategic development and the efficient transformation of azoalkenes to chiral heterocycles and α-functionalized ketone derivatives since 2010. The plausible mechanism for each reaction model is also discussed.
 Liang Wei | Liang Wei was born in Xuan Cheng, Anhui, in 1990. He received his BS degree in chemistry from Hubei University in 2012. In the same year, he joined the College of Chemistry and Molecular Sciences at Wuhan University as a graduate student in a “1+4” master-and-doctoral program and received his PhD in organic chemistry in 2017 under the guidance of Professor Chun-Jiang Wang. His research interests focus on asymmetric transition-metal catalysis and synthesis of bioactive compounds. |
 Chong Shen | Chong Shen was born in Hangzhou, Zhejiang, in 1991. He received his BS degree in chemical engineering from Zhejiang University of Technology in 2013. And then obtained his MS degree in organic chemistry from Zhejiang University of Technology in 2016 under the guidance of Professor Yixia Jia. After that, he joined the College of Chemistry and Molecular Sciences at Wuhan University for his PhD in organic chemistry in 2016 under the guidance of Professor Chun-Jiang Wang. His research interests focus on asymmetric transition-metal catalysis and synthesis of biologically active and enantiomerically enriched compounds. |
 Chun-Jiang Wang | Chun-Jiang Wang received his PhD in organic chemistry in 2003 under the guidance of Professor Min Shi at Shanghai Institute of Organic Chemistry. After that, he conducted postdoctoral research with Professor Xumu Zhang at Penn State University. In 2006, he joined the College of Chemistry and Molecular Sciences at Wuhan University as an associate professor and was promoted to full professor in 2008. His research aims at the design and synthesis of chiral ligands and organocatalysts for efficient construction of biologically active and enantiomerically enriched compounds. |
Introduction
The efficient synthesis of enantiomerically enriched nitrogen-containing heterocycles continues to be a major focus of research efforts in the modern synthetic organic community due to their remarkable activity and wide applications.1 In this context, the catalytic asymmetric cycloaddition reactions represent extremely powerful tools and impressive developments have been achieved in recent decades.2 However, in comparison with the well-established cycloaddition strategies to construct heterocycles containing one nitrogen atom, such as pyridines, piperidines and pyrroles, there are only a few methods being disclosed for the synthesis of multi-nitrogen-containing heterocyclic frameworks.3Azoalkene, which is also called enyldiazene or 1,2-diaza-1,3-butadiene, has proven to be an efficient starting material for the synthesis of various multi-nitrogen-containing heterocycles including pyrroles, pyrazoles, imidazoles, thiazoles, selenazoles, 1,2,3-thiadiazoles, 1,2,3-selenadiazoles, 1,2,3-diazaphospholes, pyridazines, pyrazines, 1,4-thiazines, 1,2,4-triazines and mixed heterocyclic systems. A solid foundation in this area has been established by O. A. Attanasi's group, which has been devoted to the non-asymmetric transformation of azoalkenes for thirty years and published two reviews about the reactivity of azoalkenes.4
However, azoalkene-involved asymmetric annulation has received less attention although the corresponding enantiomerically enriched multi-nitrogen-containing heterocycles exhibit potential applications in drug synthesis.
Recently, readily available α-halo N-acyl hydrazones, serving as the precursor of azoalkenes via base-promoted 1,4-elimination5 (Scheme 1), have been successfully employed in the construction of optical active heterocycles and considerable progress has been made since 2010. In this feature article, we highlight recent progress on asymmetric annulation of azoalkenes reported by our laboratory and others. Meanwhile, some successful racemic examples are also discussed. To calibrate the reasonable scope of this survey, this feature article is accordingly organized by the different types of reaction model.
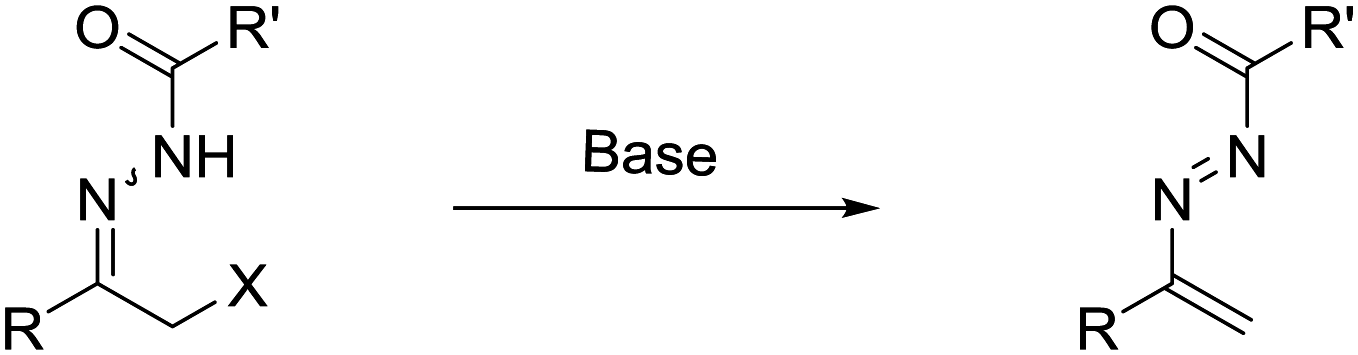 | ||
| Scheme 1 Readily-available α-halo N-acyl hydrazone serving as a precursor of azoalkenes. | ||
[4+1] Annulation of azoalkene
Dihydropyrazoles are important five-membered aza-hetero-cycles and are present in a wide range of bioactive compounds with anti-depressant, anti-cancer, anti-inflammatory, anti-bacterial, and anti-viral activities.6 Moreover, functionalized dihydropyrazoles are also of significance for the preparation of natural products and applications in asymmetric synthesis. The catalytic asymmetric formal [4+1] annulation of azoalkenes with C1 synthons provides direct access to enantiomerically enriched dihydropyrazoles.Asymmetric [4+1] annulation with sulfur ylides
In 1973, Dalla and coworkers reported the first example of the [4+1] annulation of azoalkenes and sulfur ylides to produce dihydropyrazole derivatives (Scheme 2, upside).7a In 2012, the Bolm group successfully realized the asymmetric version of this reaction with the chiral Lewis acid complex (Scheme 2, downside).7b The azoalkene is formed in situ by the base treatment of α-chlorinated hydrazone 1. The nucleophilic attack of sulfur ylide 2 at the terminal position of the in situ formed azoalkenes followed by the intramolecular cyclization with the release of dimethyl sulfide, in a stepwise fashion, completes the transformation. The chiral environment of this reaction is created by the coordination of the Cu(OTf)2/(R)-Tol-BINAP complex with the nitrogen and oxygen atoms of the in situ formed azoalkenes. A series of dihydropyrazoles 3 are produced in high yields (81–97%) with moderate to excellent enantioselectivities (42–94% ee). Some selected examples of the substrate scope are shown in Scheme 2.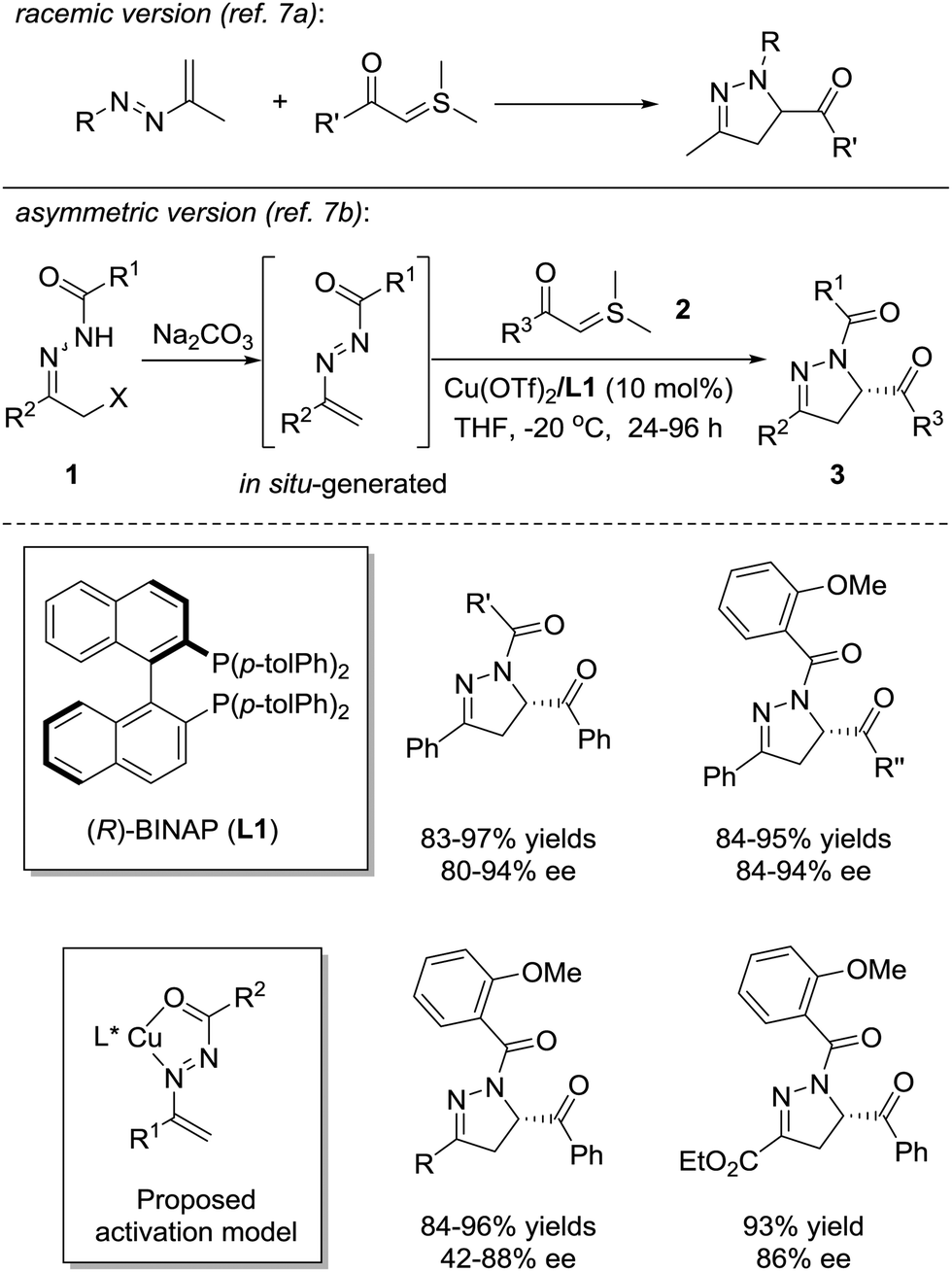 | ||
| Scheme 2 Formal [4+1] annulation of sulfur ylides with in situ generated azoalkenes. | ||
Very recently, Wang and Fang et al. reported a similar transformation using in situ-formed fluorinated sulfur ylides as C1 reactants, which provided a series of racemic 5-(trifluoromethyl)pyrazolines in good yields without any catalysts.8
[4+1] Annulation of azoalkene with diazo compounds
In 2014, the Favi group reported a formal [4+1] cycloaddition of azoalkenes and diazo compounds.9 The diazo esters 4 react with azoalkenes through the carbon as a soft nucleophilic centre and act as a synthetic equivalent of a 1,1-dipole one-carbon synthon or a carbene. Notably, under catalyst-free conditions, the dimer 7 of the in situ formed azoalkene could be isolated with a 62% yield while only a 17% yield of the desired [4+1] product 6 was obtained. Employing ZnCl2, Bi(OTf)3, InCl3, Yb(OTf)3 or Sc(OTf)3 as the Lewis acid catalyst all give dimer 7 as the major product. Switching the catalyst to copper salts reverses the ratio and CuCl2 shows the best performance, giving the desired product in a 92% yield, and the self-[4+2] annulation is completely suppressed (Scheme 3).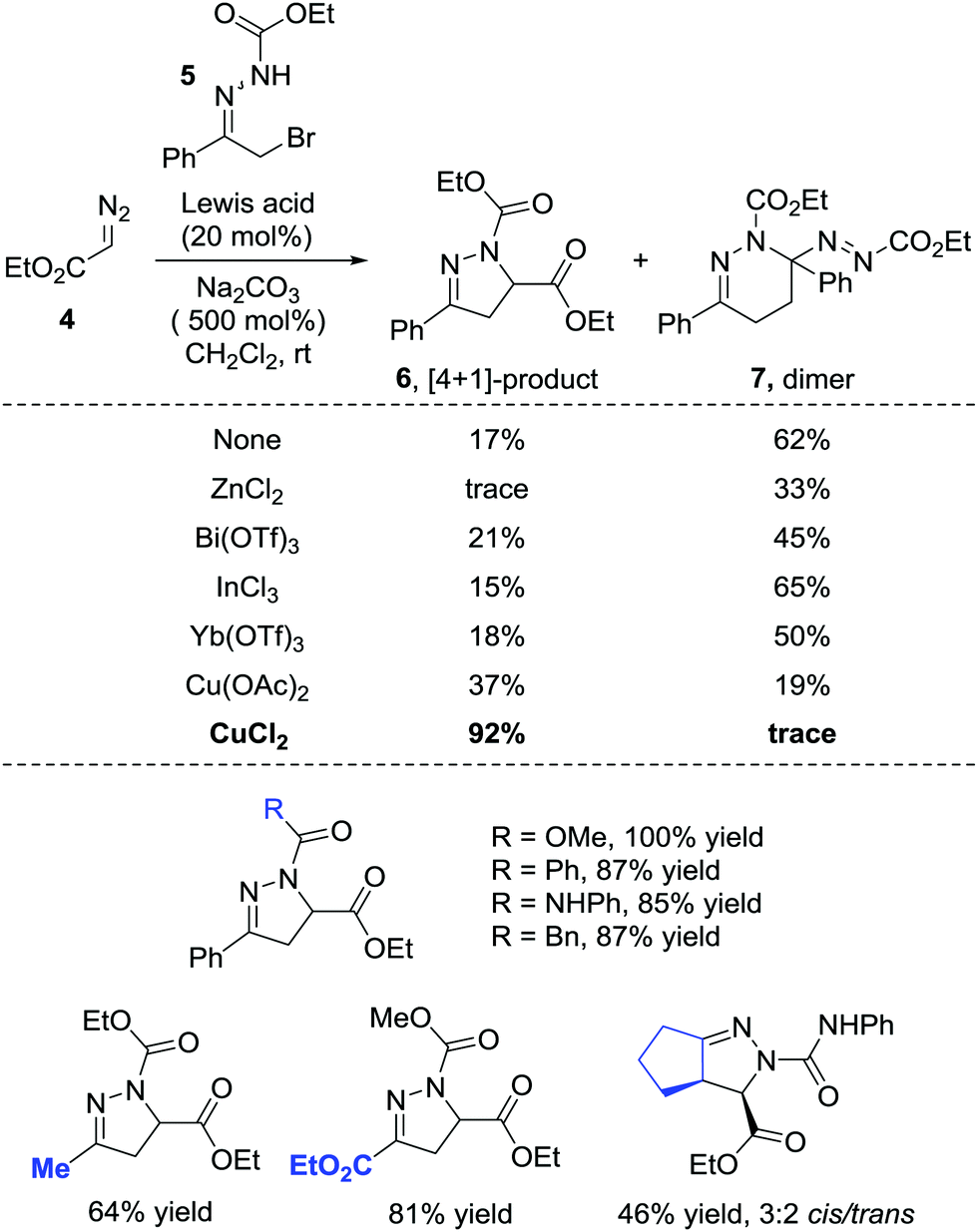 | ||
| Scheme 3 Cu(II)-Catalyzed formal [4+1] annulation of diazo compounds with in situ generated azoalkenes. | ||
Formal [4+1]/[4+2] annulation of azoalkene with maleimide
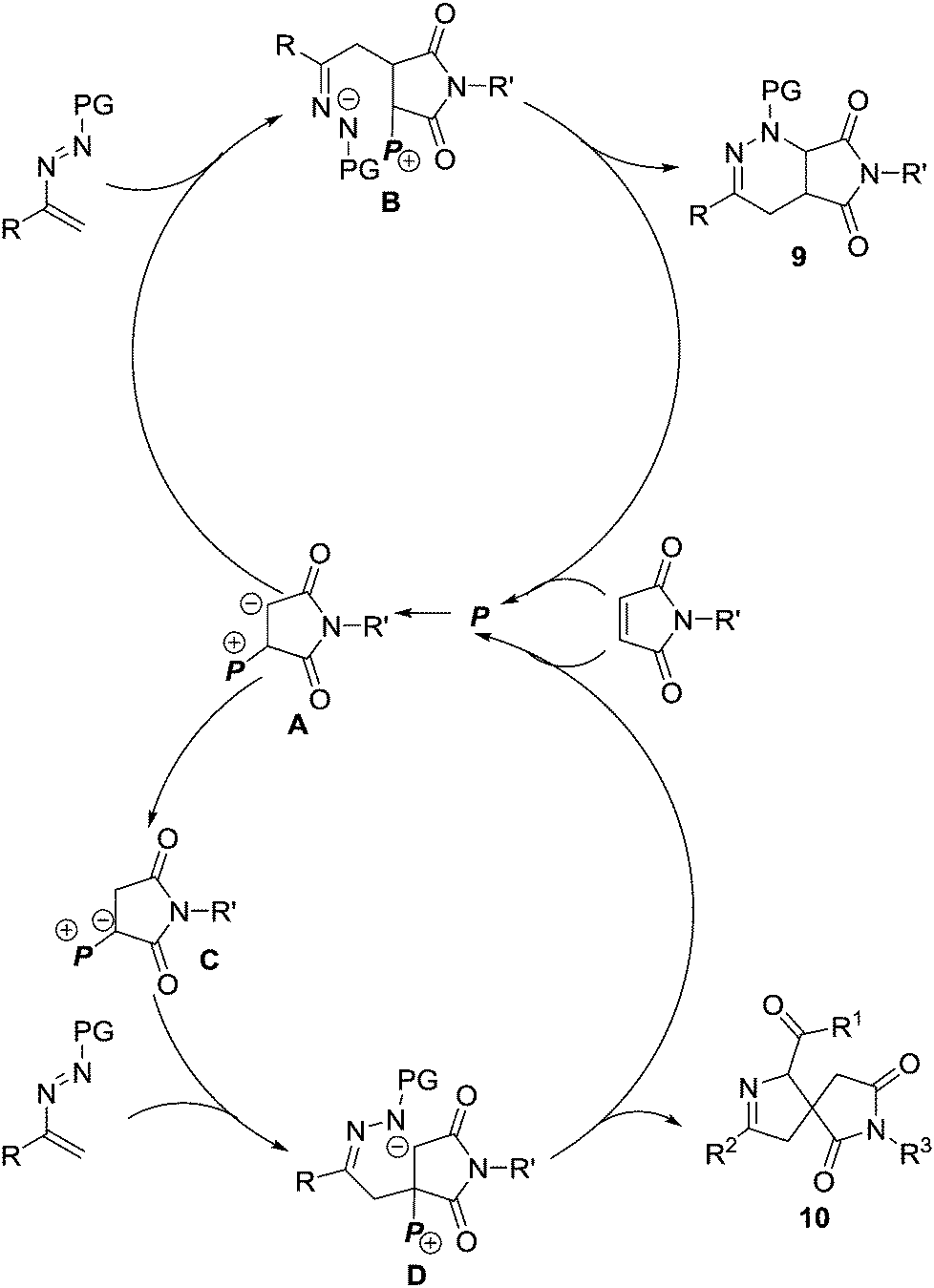
In 2017, our group reported unprecedented PPh3-mediated [4+2]/[4+1] annulations of maleimides 8 with azoalkenes, in which maleimide served as C2 and C1 synthons. The fused tetrahydropyridine/pyrrolidinedione 9 and spiro-dihydropyrazole/pyrrolidinedione derivatives 10 were achieved in good yields from the same set of starting materials under different reaction conditions.10 The catalyst and solvent screening indicated that using 20 mol% of PPh3 as the catalyst and 1,2-dichloroethane as the solvent, the reaction proceeded exclusively via a [4+2] annulation pathway. Only [4+1] products were isolated when this reaction was performed with a stoichiometric amount of PPh3 in acetone.11 Meanwhile, temperature screening shows that increasing the reaction temperature is beneficial for the [4+2] pathway (Scheme 4).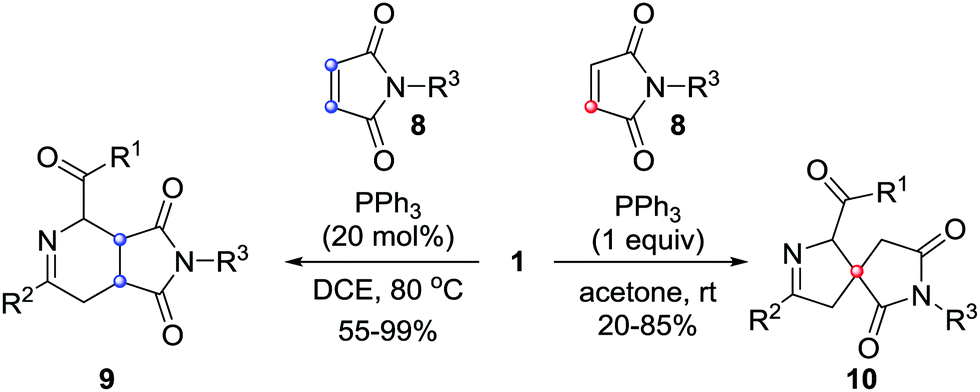 | ||
| Scheme 4 PPh3-Mediated formal [4+1]/[4+2] annulation of maleimide with in situ generated azoalkenes. | ||
We proposed a possible reaction mechanism to rationalize the PPh3-mediated [4+2]/[4+1] annulations of maleimides and azoalkenes (Scheme 5). Both pathways begin with the nucleophilic attack of PPh3 (P) on the electron-deficient C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in maleimide generating the zwitterionic intermediate A. In the lower polar solvent 1,2-dichloroethane, intermediate A acts as a carbon-based nucleophile and directly attacks the electron-deficient azoalkene to form zwitterionic species B, which is followed by the intramolecular cyclization to furnish the fused product 9 and regenerate PPh3. When using the more polar solvent acetone instead of 1,2-dichloroethane with a stoichiometric amount of PPh3, the initially generated zwitterionic intermediate A is efficiently converted to a more stable phosphorus ylide C, which is believed to be facilitated by acetone-aided 1,2-proton transfer. Phosphorus ylide C can serve as a 1,1-dipole C1 synthon and react with the azoalkene in a stepwise formal [4+1] pathway, affording the spiro heterocyclic product 10.
C bond in maleimide generating the zwitterionic intermediate A. In the lower polar solvent 1,2-dichloroethane, intermediate A acts as a carbon-based nucleophile and directly attacks the electron-deficient azoalkene to form zwitterionic species B, which is followed by the intramolecular cyclization to furnish the fused product 9 and regenerate PPh3. When using the more polar solvent acetone instead of 1,2-dichloroethane with a stoichiometric amount of PPh3, the initially generated zwitterionic intermediate A is efficiently converted to a more stable phosphorus ylide C, which is believed to be facilitated by acetone-aided 1,2-proton transfer. Phosphorus ylide C can serve as a 1,1-dipole C1 synthon and react with the azoalkene in a stepwise formal [4+1] pathway, affording the spiro heterocyclic product 10.
 | ||
| Scheme 5 Proposed mechanism for the PPh3-mediated formal [4+1]/[4+2] annulation of maleimides with in situ generated azoalkenes. | ||
[4+2] Annulation of azoalkene
Pyridazines and hydropyridazines are versatile structural motifs in a number of natural products (e.g., pyridazomycin and azamerone) and pharmaceutically active compounds such as nonsteroidal progesterone receptor regulators, neuro-transmitter inhibitors, and influenza neuraminidase inhibitors.12 The known synthetic strategies for tetrahydropyridazines are limited to a few special substrates, and few general methods exist for their synthesis.5 Strategically, the newly discovered inverse electron-demand aza-Diels–Alder reaction of in situ formed azoalkenes with dienophiles is arguably one of the most powerful and atom-/step-economic approaches. Although many examples of azoalkene-involved [4+2] annulation have been reported, most of them are racemic or have moderate levels of enantioselectivities.13 Notably, the in situ formed azoalkenes are a highly reactive species, and the uncatalysed background reaction was observed in many cases, which constitutes the main obstacle to realize the corresponding asymmetric transformation.Formal [4+2] annulation of azoalkene with aldehyde and arylacetic acids
In 2010, the Pitacco group reported the first example of the formal [4+2] annulation of azoalkenes 11 with phenylacetaldehydes 12 catalysed by L-proline or (S)-(+)-1-(2-pyrrolidinylmethyl)-pyrrolidine. However, these two amines did not show sufficient catalytic reactivity or stereocontrol ability, and the corresponding 1,4-dihydropyridazines 13 were obtained in 15–87% yields and 25–78% ee with relatively long reaction time (4–10 d) (Scheme 6).14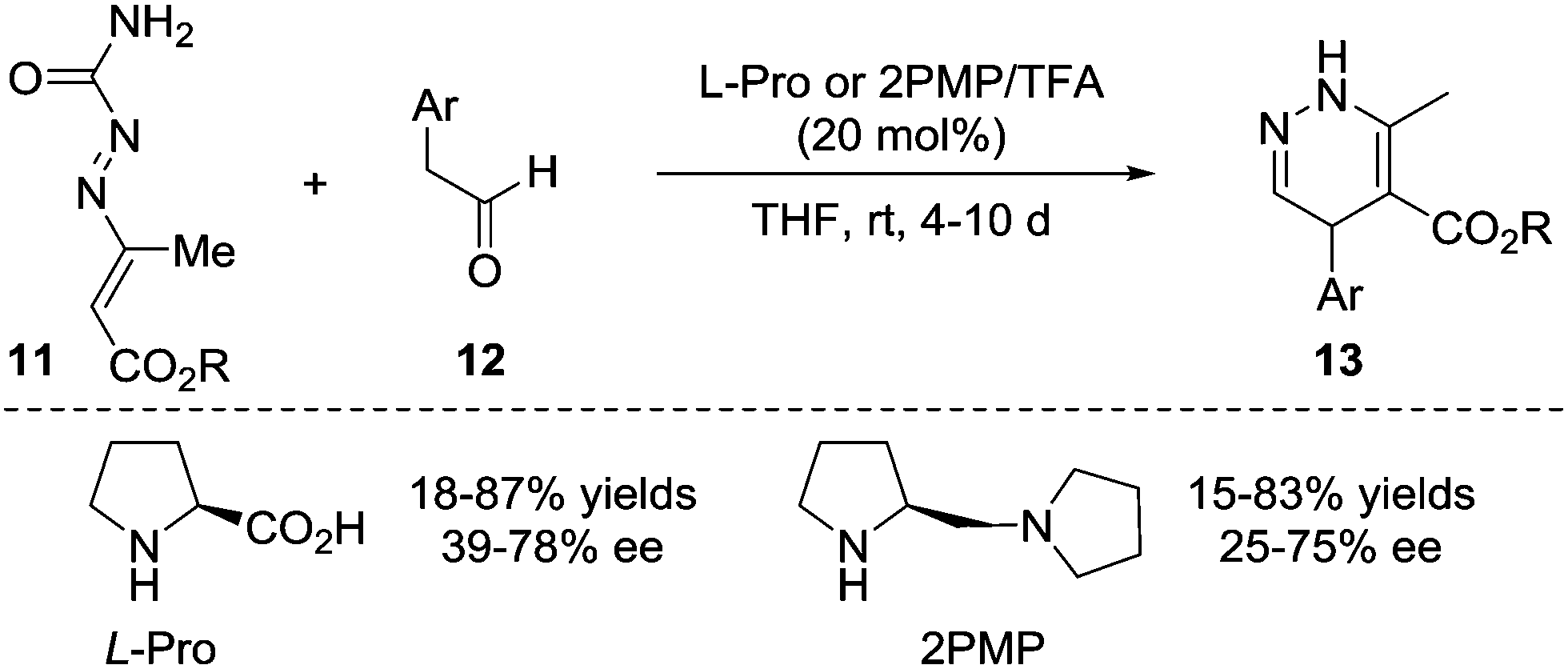 | ||
| Scheme 6 Amine catalyzed formal [4+2] annulation of aldehydes with azoalkenes. | ||
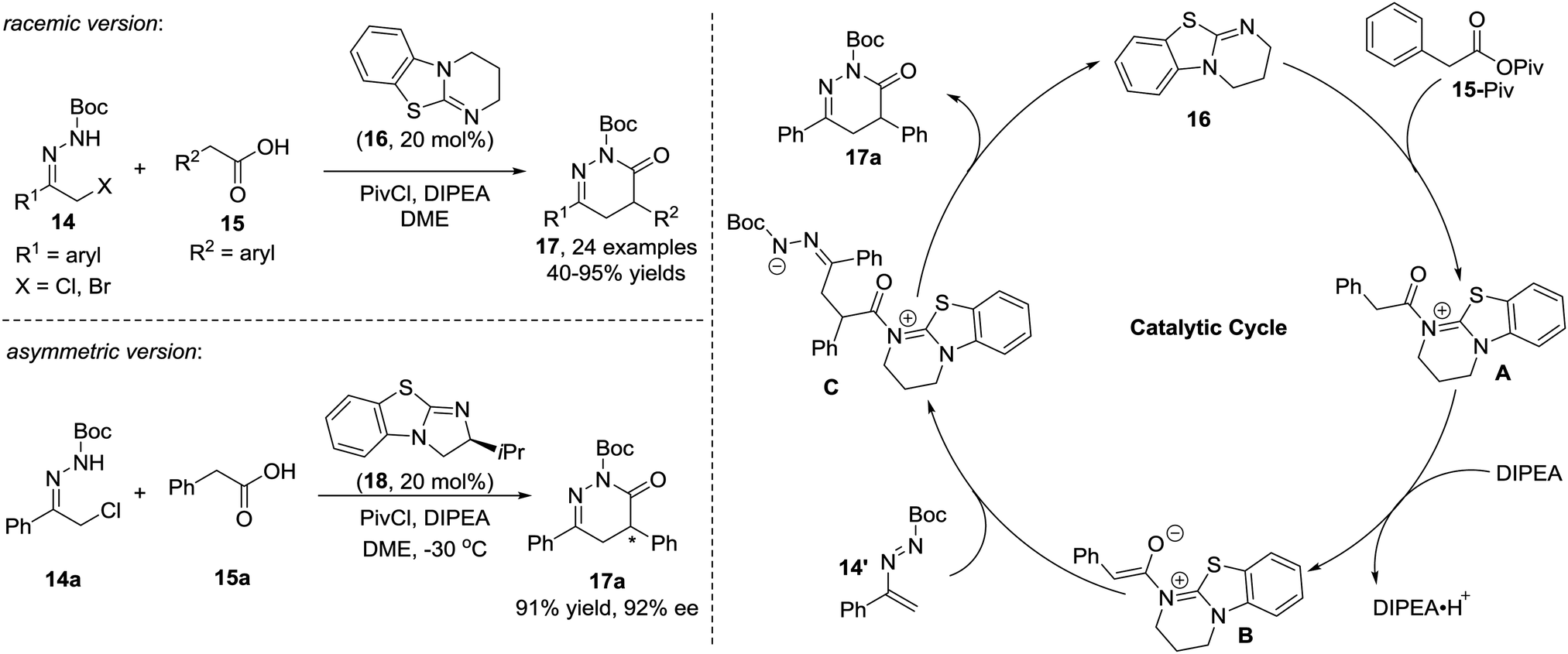
Yao et al. in 2015 reported an isothiourea 16 catalysed formal [4+2] cycloaddition of azoalkenes 14 with arylacetic acids 15 in a racemic version.15 The reaction mechanism is proposed to involve the initial formation of intermediate A between isothiourea 16 and anhydride 15-Piv which is in situ generated from 15 and PivCl. The deprotonation of A under basic conditions affords ammonium enolate B. Nucleophilic attack by the enolate at the terminal position of the in situ formed azoalkene 14′ followed by intramolecular lactamization produces the corresponding 4,5-dihydropyridazin-3(2H)-one 17 and regenerates the catalyst. The authors also tested the asymmetric version of this reaction with one example using (S)-BTM as the chiral catalyst. Gratifyingly, the product 17a could be achieved in a 91% yield with a 92% ee (Scheme 7).
 | ||
| Scheme 7 Isothiourea catalyzed formal [4+2] cycloaddition of azoalkenes with arylacetic acids and the proposed mechanism. | ||
[4+2] Annulation of azoalkene with electron-rich alkenes
In 2013, Xiao et al. reported that Lewis acids catalysed the inverse electron-demand aza-Diels–Alder (IEDDA) reaction of azoalkenes with enol ethers 19. Using a catalytic amount of the chiral Cu(OTf)2/bisoxazoline(L2) complex, the reaction allows rapid access to tetrahydropyridazine derivatives 20 in good efficiency with moderate to excellent enantioselectivities (14–90% ee) (Scheme 8, upside).16 In 2015, our group also reported a similar transformation using the Cu(I)/tBu-phosferrox(L3) complex as the catalyst.17 The corresponding products were isolated in high yields with generally excellent enantioselectivities (Scheme 8, downside).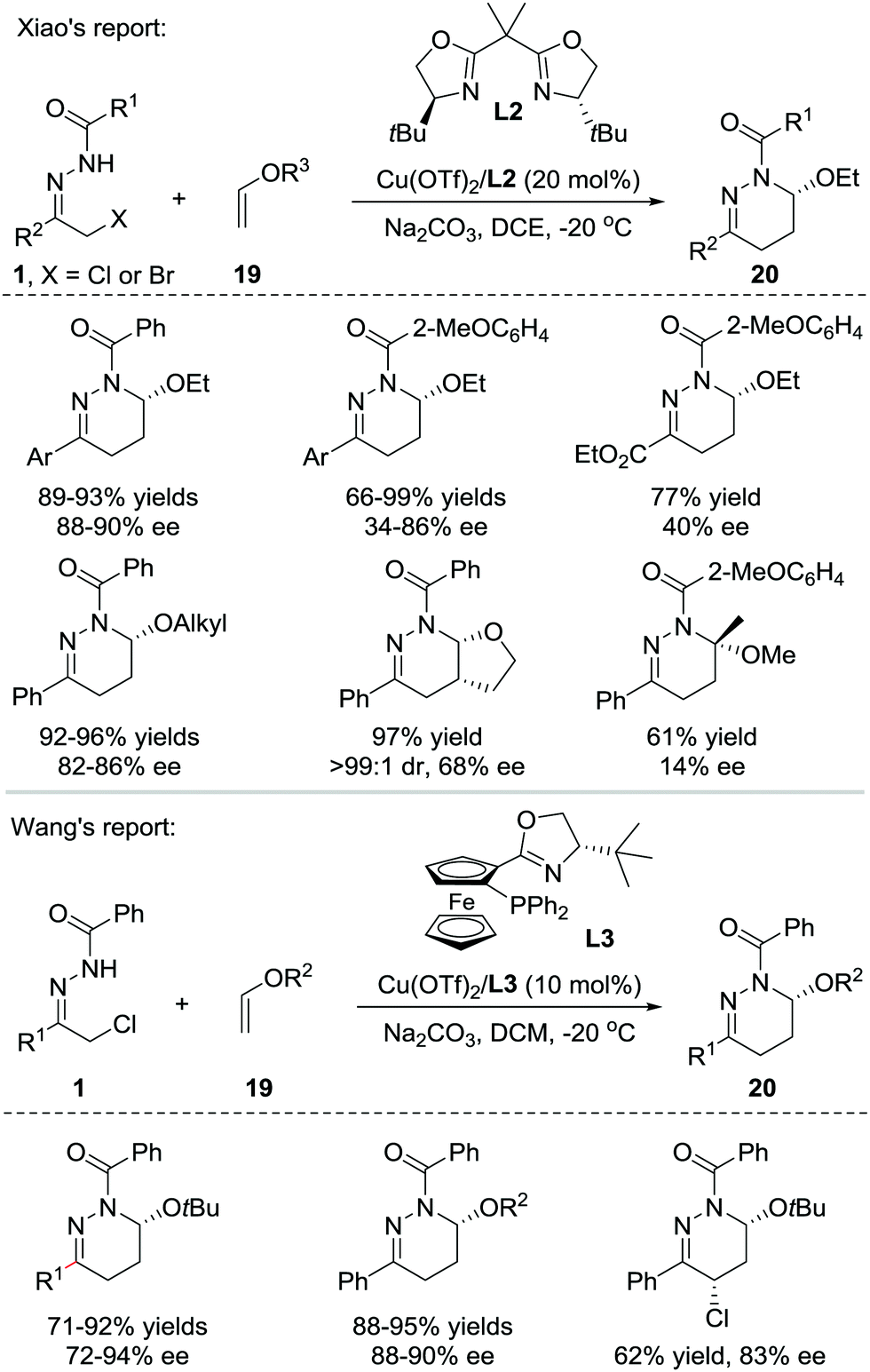 | ||
| Scheme 8 Copper complex catalyzed IEDDA reaction of azoalkene with enol ethers. | ||
Recently, much synthetic effort has been made to develop efficient strategies for the dearomative annulation of indoles to prepare biologically important [2,3]-fused indoline heterocycles.18 In this context, the catalytic asymmetric intra-19a–g and intermolecular19h–m C2,C3-annulation of indoles, has become one of the most direct and diversity-oriented methods20 for the construction of optically active [2,3]-fused indolines. In 2014, our group successfully developed the first catalytic asymmetric dearomative IEDDA reaction of indoles with 21 azoalkenes catalysed by a Cu(I)/tBu-phosferrox(L3) complex.21 The catalyst system shows excellent efficiency and asymmetric induction ability with satisfactory tolerance to various functional groups, delivering a series of [2,3]-fused indoline tetrahydropyridazines 22 in 80–95% yield with 78–99% ee. Remarkably, the branched α,α-dichloro hydrazone 1′ is also a suitable azoalkene precursor, affording the heterocycle 22′ bearing three contiguous stereogenic centers (Scheme 9).
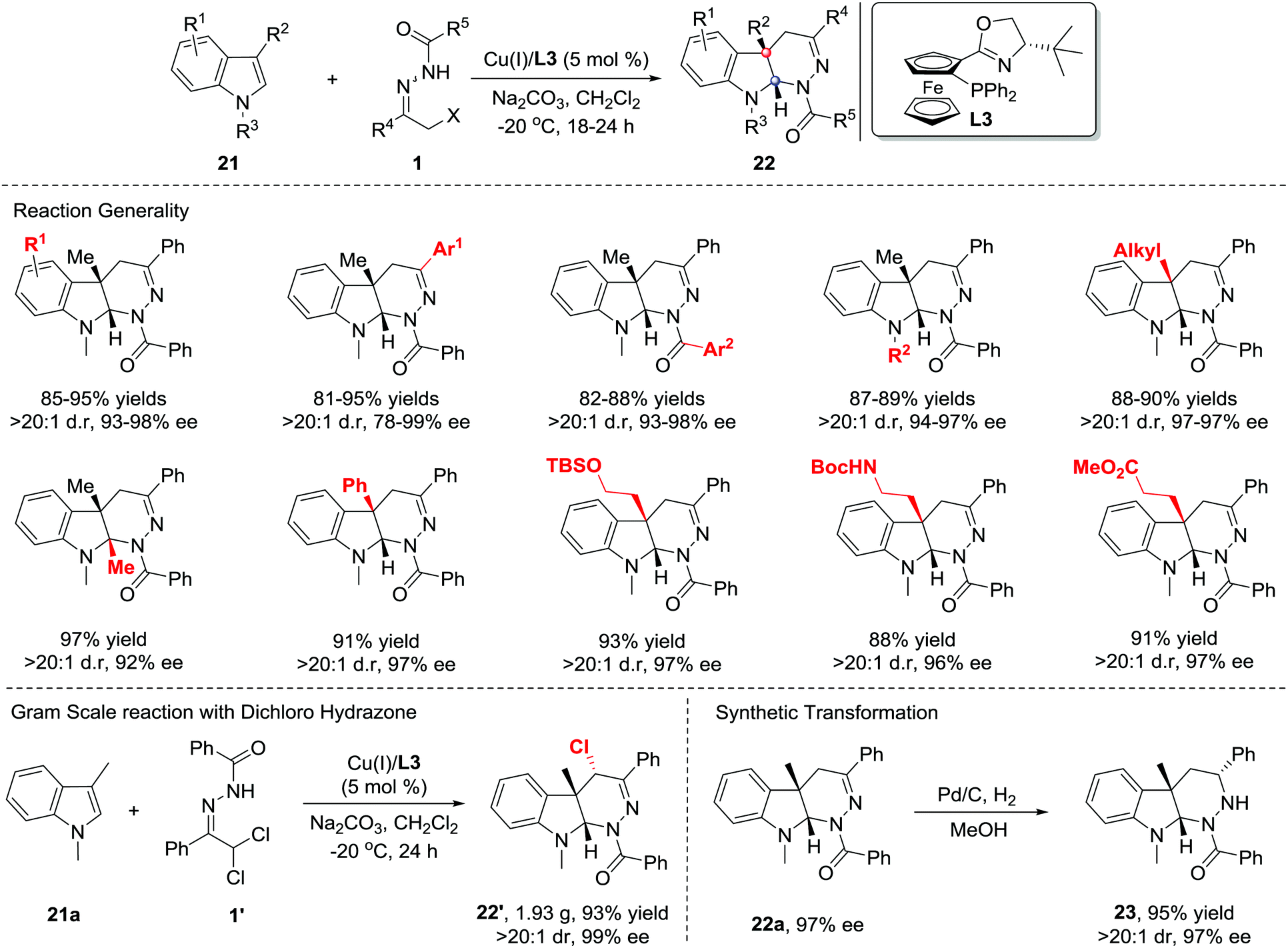 | ||
| Scheme 9 Cu(II)/tBu-phosferrox catalyzed inverse-electron-demand Diels–Alder reaction of azoalkene with indoles. | ||
2-Siloxyfurans, which are commonly recognized as readily accessible nucleophilic synthons of the γ-anion of 2(5H)-furanone, have been widely used in vinylogous Mukaiyama–aldol, Mukaiyama–Michael, and Mukaiyama–Mannich type additions to prepare butenolide derivatives.22 Considering that butenolide is a potential Michael acceptor, we envisioned that 2-silyloxyfurans might sequentially react as a nucleophile and an electrophile in the domino reaction, giving rise to fused butyrolactones, which are core structures in various natural products.23 In 2015, we developed a formal [4+2] annulation of 2-silyloxyfuran 24 with azoalkenes through a cascade vinylogous Mukaiyama 1,6-Micheal/aza-Micheal addition process.24 The challenge of this reaction is the strong uncatalysed background reaction. The Cu(I)/(S,Sp)-tBu-phosferrox complex we used before only gives moderate stereoselectivity and the Cu(OTf)2/tBu-box(L2) complex is found to be superior after screening of a large number of combinations of metal salts and chiral ligands. The protic additive hexafluoroisopropanol (HFIPA) is beneficial for improving the chemical yield.25 Under the optimized reaction conditions, azoalkene precursors with an array of (hetero)aryl substituents react smoothly with 2-silyloxyfuran to the desired products in high yields with excellent enantioselectivities and exclusive diastereoselectivities. The 2- and 5-substituted 2-silyloxyfurans are also compatible with this reaction. In addition, pyrrole-based dienoxysilane was then evaluated, and the fused butyrolactams were obtained in satisfactory yields with excellent stereoselectivity control. Unfortunately, the alkyl-substituted hydrazone was not viable, affording the product as a racemate probably due to the overwhelmingly disadvantageous background reaction (Scheme 10).
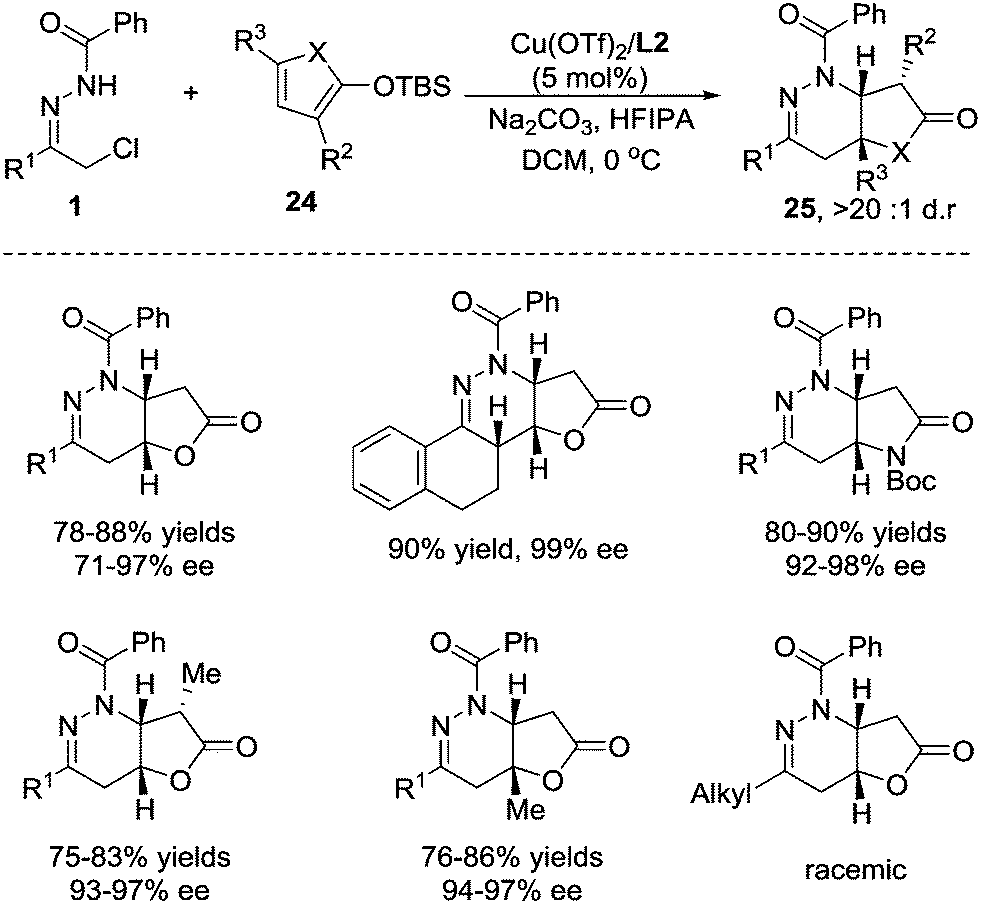 | ||
| Scheme 10 Cu(II)/tBu-Box catalyzed formal [4+2] annulation of azoalkene with 2-silyloxyfuran. | ||
The investigation of carbon isotope effects for the current reaction between TBSOF and azoalkene indicates that only a noticeable carbon isotope effect was detected on the carbon at the 5-position (13Crecovered/13Cvirgin = 1.029, average of three runs), revealing that the first vinylogous Mukaiyama 1,6-Michael addition is the rate-determining step.26 Thus, we believe that this annulation proceeds through a stepwise pathway that is not a concerted [4+2] cycloaddition, followed by sequential desilylation (Scheme 11).
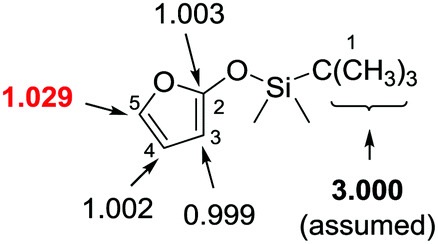 | ||
| Scheme 11 Carbon isotope effects (R/R0) calculated for TBSOF furan. The three methyl carbons of the tBu group (in bold) were taken as the internal standard. | ||
When we attempted to further extend the dearomative azoalkene-involved inverse-electron-demand Diels–Alder (IEDDA) reaction to challenge the 2-MeO-substituted furans 26, which were generally utilized as 4π participants in the Diels–Alder reaction and [4+3] cycloaddition,27 an unprecedented multicomponent cascade IEDDA/nucleophilic addition/ring-opening process was realized serendipitously.28 The anticipated bicyclic heterocycle was not observed; instead, the cycloadduct was further attacked by H2O, followed by tetrahydrofuran ring-opening, leading to unexpected tetrahydropyridazine derivatives 27 bearing a γ-hydroxyl ester moiety. The existence of intermediate 28 was confirmed by trapping other nucleophiles, such as MeOH and EtOH, instead of water. Compound 30 was isolated as a single isomer, further indicating that the nucleophilic addition to intermediate 28 is stereospecific. The ligand we used in the reaction of azoalkenes and 2-silyloxyfurans also exhibited excellent stereocontrol ability in this transformation using Cu(MeCN)4BF4 as the metal salt. Various substituted hydrazone and 2-alkoxy furans were evaluated, producing the corresponding products with satisfactory results. Moreover, this method provides a more atom-economic way to fuse γ-butyrolactone 25a compared with our previous work via the base-promoted intramolecular lactonization (Schemes 12 and 13).
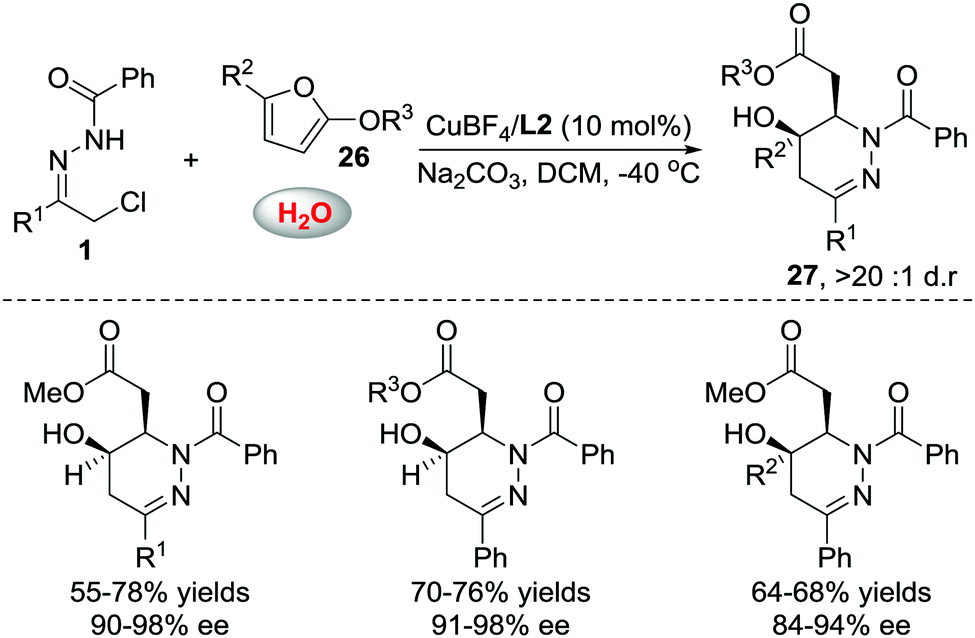 | ||
| Scheme 12 Cu(I)L2 catalyzed multicomponent cascade inverse electron-demand aza-Diels–Alder/nucleophilic addition/ring opening reaction. | ||
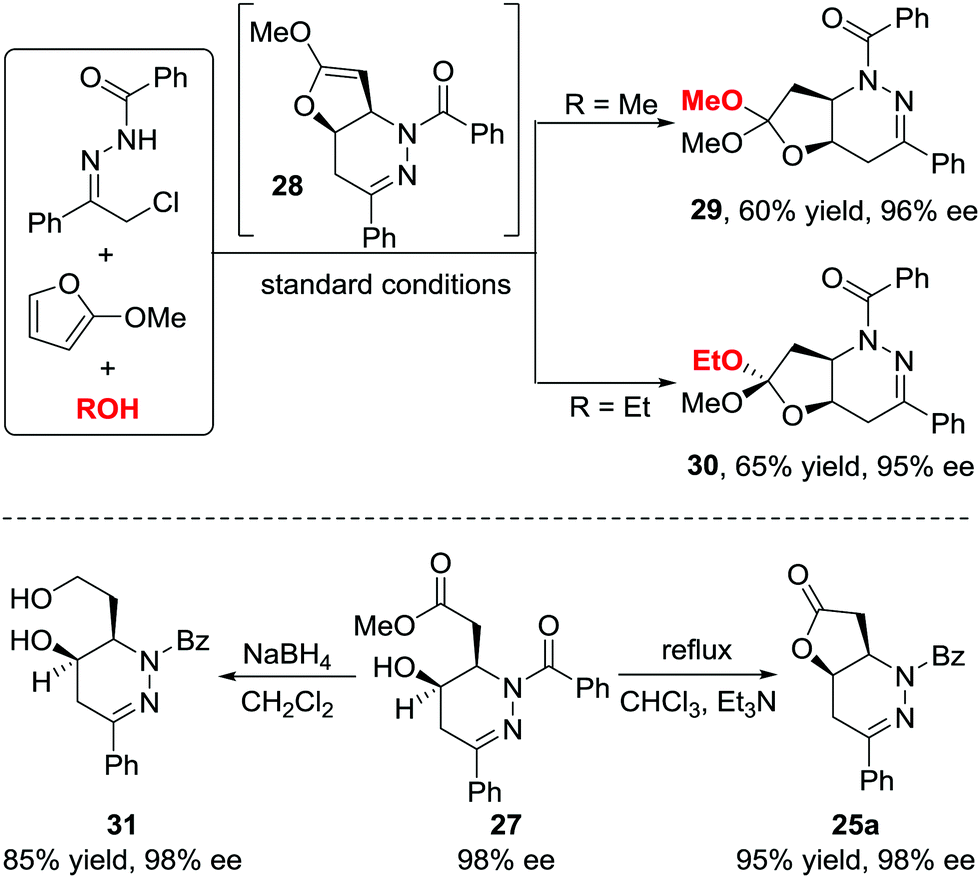 | ||
| Scheme 13 Control experiments to capture the intermediate and synthetic transformation. | ||
Enoldiazoacetates are widely used as electron-rich alkenes or 1,3-dipoles in cycloaddition reactions.29 Xu and Doyle recently realized catalyst-dependent [4+2]-cycloaddition reactions of azoalkenes with enol diazoacetates 32.30 When the reaction between α-halo hydrazone and enol diazoacetates was conducted under basic conditions without any catalysts, tetrahydropyridazinyl-substituted diazoacetates 34 were produced as the major products. In contrast, when Rh2(OAc)4 was employed as the catalyst, the enol diazoacetates underwent Rh2(OAc)4-catalysed dinitrogen extrusion to generate the donor–acceptor cyclopropenes 33 first, followed by [4+2]-cycloaddition with azoalkenes to yield bicyclo[4.1.0]tetrahydropyridazines 35 (Scheme 14).
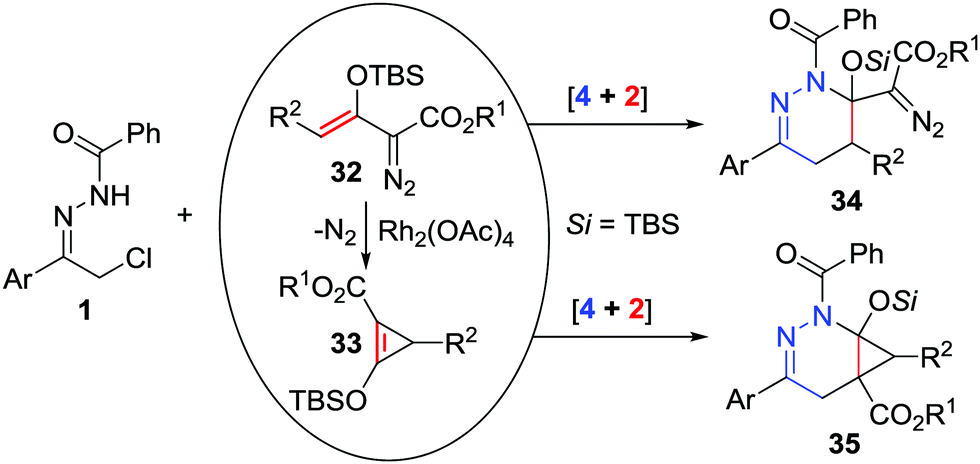 | ||
| Scheme 14 The regioselective [4+2] annulation of enoldiazoacetate with azoalkenes. | ||
[4+2] Annulation of azoalkene with electron-neutral alkenes
In addition to the electron-rich alkenes, the electron-neutral alkenes are also suitable dienophiles in the IEDDA reaction due to the high reactivity of azoalkene. In 2015, Luo et al. reported a catalyst-free [4+2] cycloaddition of in situ generated azoalkenes and simple olefins.31 The reaction proceeds smoothly under basic conditions in the absence of any catalysts. Various olefins, such as ethylene, styrene, norbornene and cyclic diene are also tolerated in this protocol, giving the desired products 36 in good yields (Scheme 15).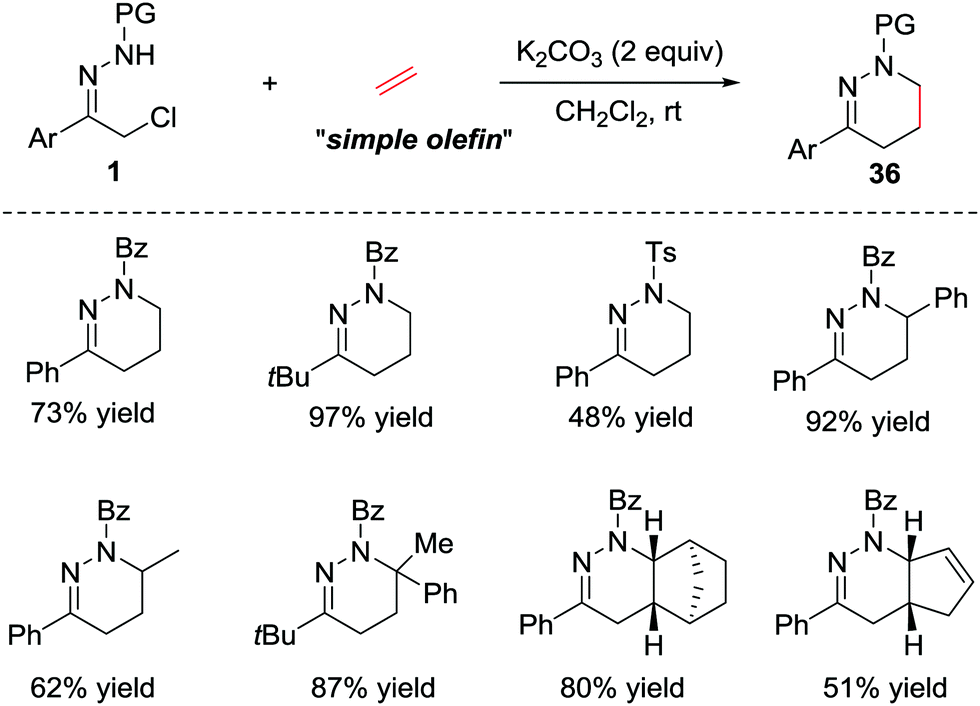 | ||
| Scheme 15 Base-promoted inverse electron-demand aza-Diels–Alder reaction of azoalkenes with simple olefins. | ||
Although Luo et al. have proven that the electron-neutral alkenes are viable substrates to react with azoalkenes, the asymmetric version of this transformation with high stereoselectivity remains elusive. Such a goal has been reached recently by our group. In 2017, we reported an unprecedented CuOTf/tBu-Box complex catalysed asymmetric IEDDA reaction of azoalkenes and 5-silylcyclopentadienes 37 in high yields with excellent enantioselectivities.5 The application of a desymmetrization strategy allowed us to construct bicyclic tetrahydropyridazine derivatives 38 bearing a unique α-chiral silane motif (Scheme 16).32
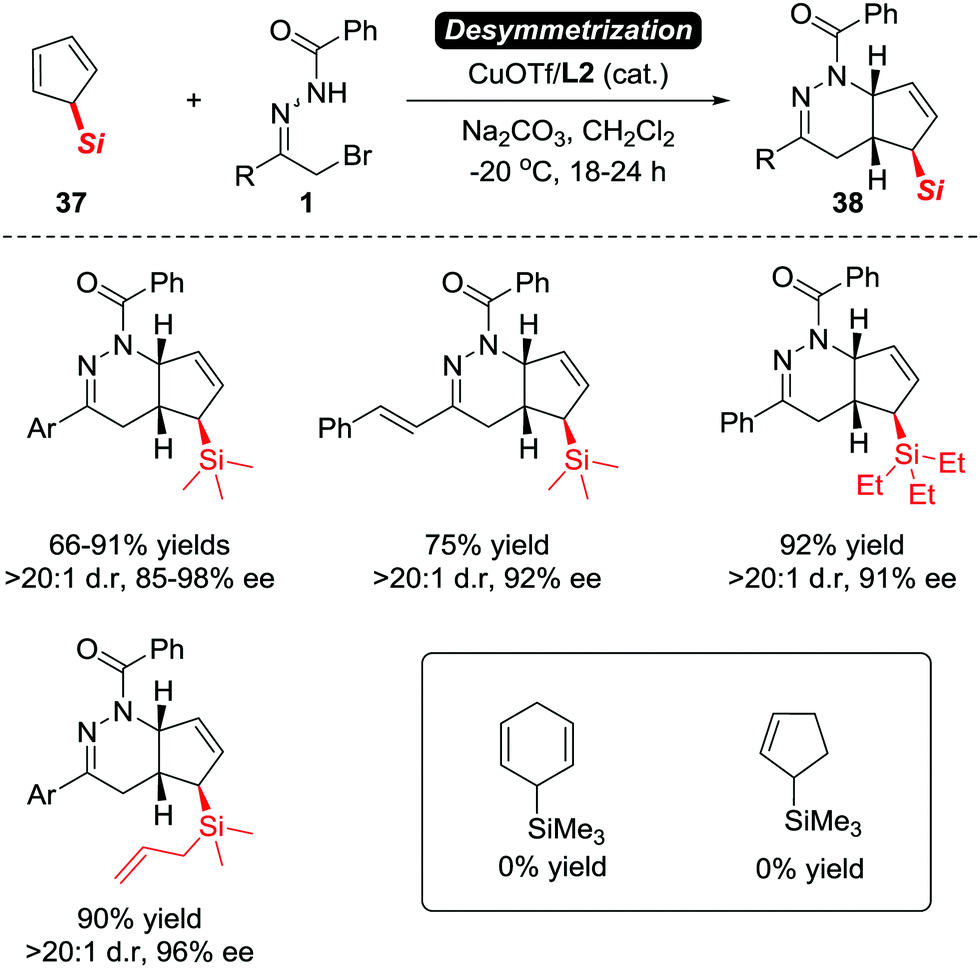 | ||
| Scheme 16 Cu(I)/L2 catalyzed asymmetric [4+2] annulation of 5-silyl cyclopentadienes with azoalkenes and its limitations. | ||
When evaluating the generality of this protocol, we noticed that unconjugated dienes, such as 1-trimethylsilyl-2,5-cyclohexadiene and 1-trimethylsilyl cyclopent-2-ene, failed to react with the azoalkenes. Considering that the cyclopentadiene derivatives are able to serve as 4π- or 2π-participants in the Diels–Alder reaction, two different possible reaction pathways are proposed: the normal-electron-demand Diels–Alder reaction (NEDDA) followed by [3,3]-sigmatropic rearrangement (pathway a), or the inverse-electron-demand Diels–Alder reaction (IEDDA) (pathway b). The computational studies with or without a catalyst and both calculation results rule out the possibility of pathway a. The calculation also indicates that the conjugation structure of cyclopentadiene is essential for this transformation because the interactions of orbitals through space lowered the energy barrier of the endo-oriented transition state (Scheme 17).33
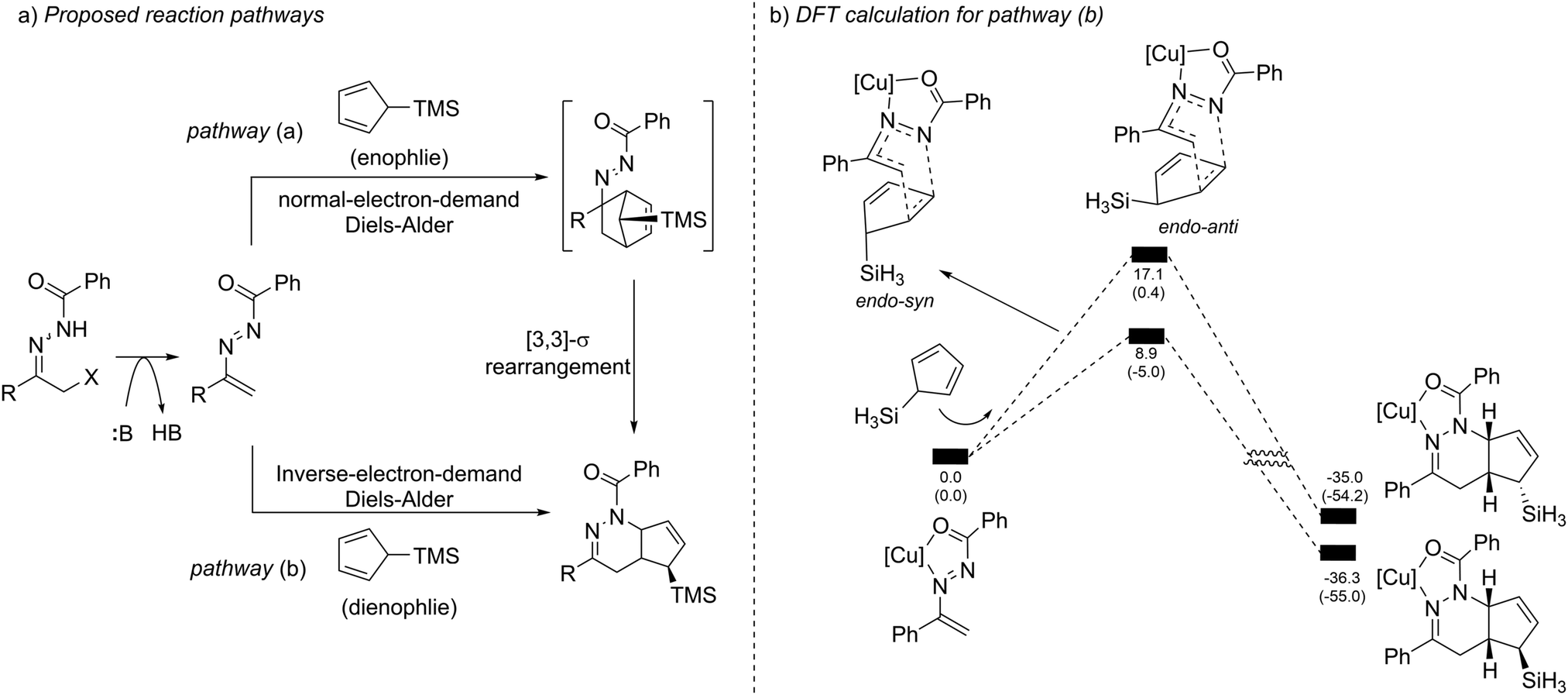 | ||
| Scheme 17 Proposed reaction pathways and computational studies. | ||
Later, we further extended the reaction to cyclopentadiene derived fulvenes 39. Compared with simple olefin and diene, fulvenes represent a more challenging reaction partner because of their capability to serve as 2π, 4π, and 6π components in various cycloadditions.34 Fortunately, the catalyst system we developed in the reaction between azoalkenes and 5-silylcyclopentadienes is also suitable here. The desired products 40 are obtained as single regio- and diastereoisomers in good yield with 92–99% ee (Scheme 18).35
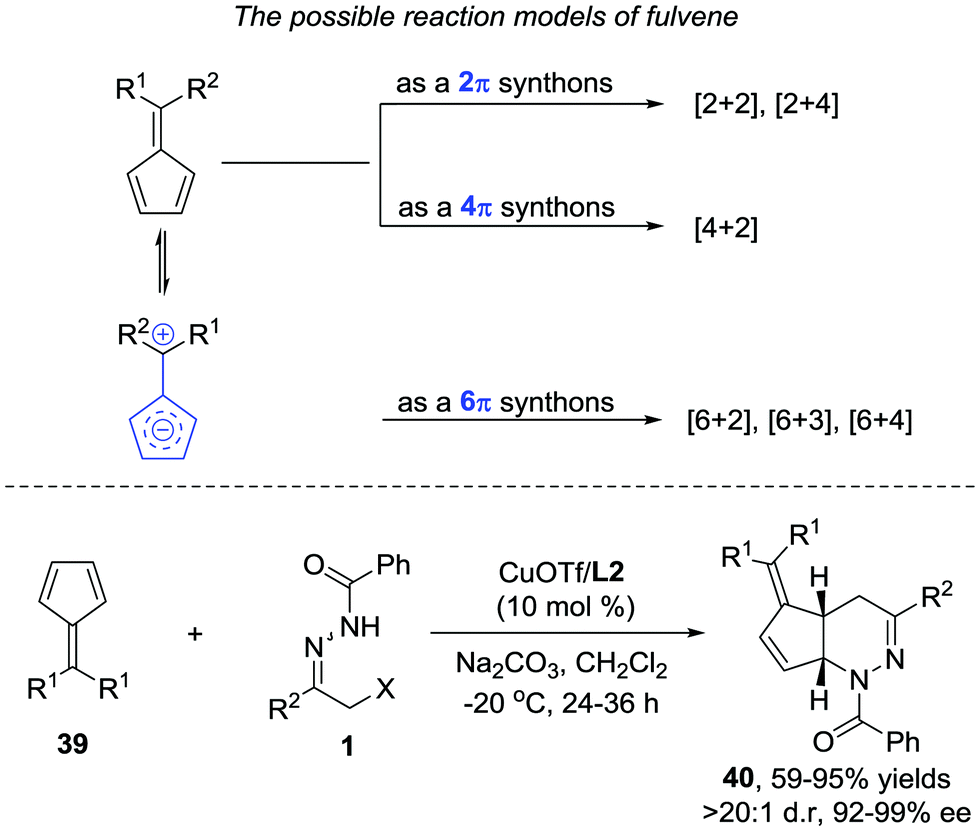 | ||
| Scheme 18 Cu(I)/tBu-Box catalyzed [4+2] annulation of azoalkenes with fulvenes. | ||
[4+3] Annulation of azoalkene
Seven-membered carbocycles and heterocycles, which are widely observed in many biologically active natural products and clinical pharmaceuticals, have attracted much attention in the development of efficient synthetic strategies.36 However, compared with the well-established methodologies for the construction of medium-sized rings (five- and six-membered), such as Diels–Alder reaction and 1,3-dipolar cycloaddition, it is relatively difficult to achieve seven-membered ring compounds, especially for seven-membered heterocycles. Representative synthetic strategies for seven-membered carbocycles include [4+3] cycloaddition, [5+2] cycloaddition, and ring-closing metathesis.27,37 In sharp contrast, methods to construct nitrogen- and multi-nitrogen-containing seven-membered heterocycles are rare. The recently developed [4+3] annulations of azoalkenes have been recognized as a straightforward protocol for the synthesis of structurally diversified seven-membered heterocycles.NHC-catalysed [4+3] annulation of azoalkene with enals
The Breslow intermediates generated from enals, which are frequently employed as C3 participants, have been well studied in NHC-catalysed [3+n] annulations.38 In 2014, Glorius et al. reported an NHC-catalysed regio- and enantioselective formal [4+3] annulation reaction between enals 41 and azoalkenes, producing the biologically important 1,2-diazepine derivatives in good yields with excellent enantioselectivities (often 99% ee).39 This reaction represents the first example of utilizing azoalkenes for the asymmetric synthesis of seven-membered heterocycles. Interestingly, the authors noticed that modifying the standard NHC catalyst could switch the reactivity towards a formal [4+1] annulation to synthesize functionalized pyrazoles. A reasonable mechanism for the regioselective umpolung annulations is proposed. As documented in Bode's study,40 an NHC catalyst with different electronics and steric demands could affect the reactive NHC-intermediate. In this reaction, controlling the competing homoenolate/acyl anion reactivity is the key factor for chemo- and regioselectivity. Here, N-Mes containing NHC was used to realize selective homoenolate reactivity and preferentially afford [4+3] cycloadducts 42, while seemingly less electron-rich N-2,6-(OMe)2 containing NHC was preferred for the enal acyl anion reaction pathway to give [4+1] annulation products 43 (Scheme 19).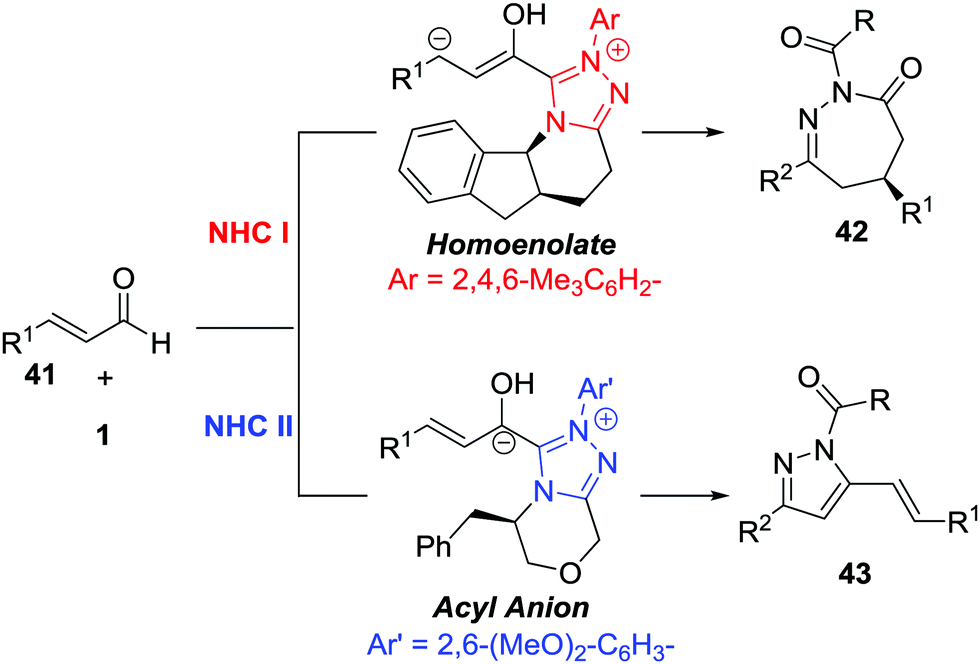 | ||
| Scheme 19 NHC-Catalyzed regioselective [4+3]/[4+1] annulation of azoalkenes with enals. | ||
Very recently, the Enders group disclosed an NHC catalysed [4+3] annulation of isatin-derived enal 44 with 4-atom components including in situ generated aza-o-quinone methides, azoalkenes, and nitrosoalkenes.41 The NHC-based homoenolate 3C components derived from enal 44 reacted smoothly with these (aza)diene compounds, affording a series of seven-membered spiro-N-heterocycles in high yields and excellent enantioselectivities. α-Chloro N-tosyl hydrazones bearing substituted-aryl, heteroaryl or alkyl groups were all found to be viable azoalkene precursors for this transformation. The Z/E geometry of the isatin-derived enal has virtually no effect on the reaction with azoalkene in yield and enantioselectivity according to the control experiments. A possible catalytic cycle is proposed for this transformation. The addition of the NHC to the isatin-derived enal generates the Breslow intermediates, which undergo conjugate addition of the aza-o-quinone methide or azoalkene to construct a new C–C bond and quaternary stereocenter. The resulting acyl azolium species then proceeds via lactamization to release the NHC catalyst and produce the desired seven-membered spiro-N-heterocycles 45 (Scheme 20).
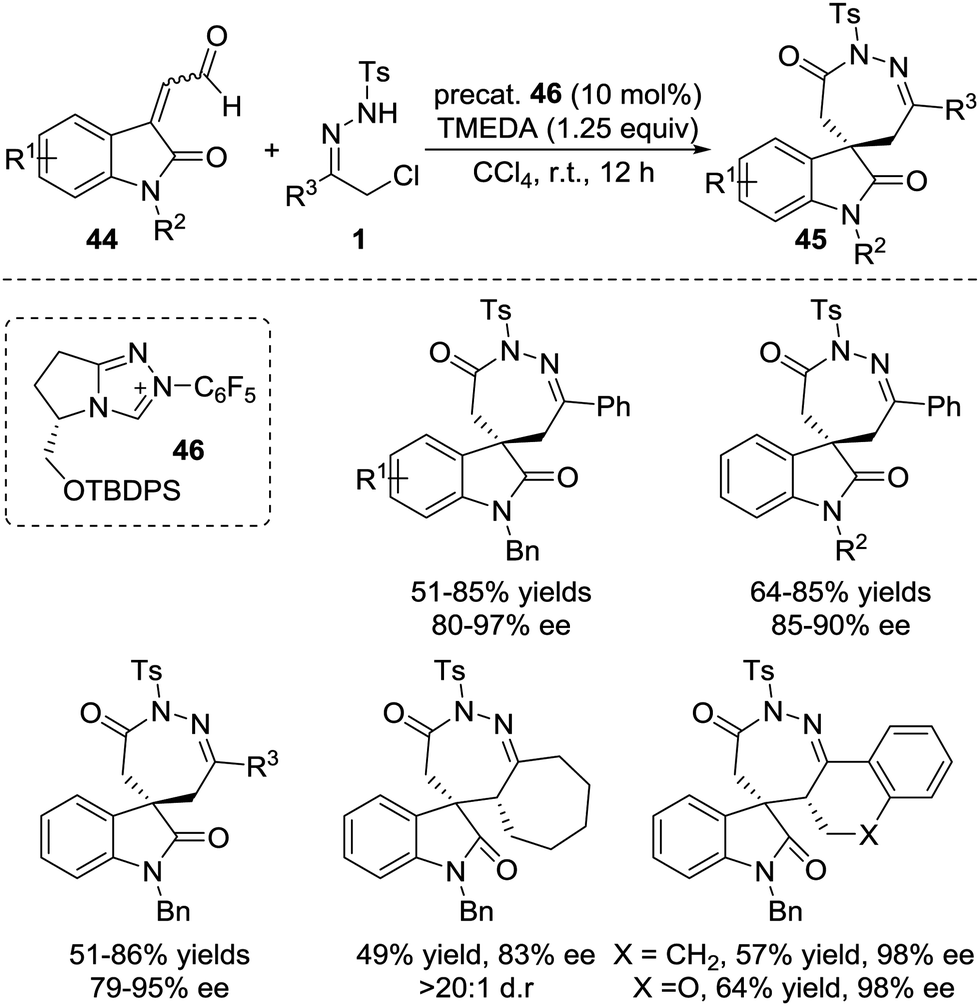 | ||
| Scheme 20 NHC-Catalyzed [4+3] annulation of azoalkenes with isatin-derived enal. | ||
Lewis acid catalysed [4+3] annulation of azoalkene with 1,3-dipoles
1,3-Dipoles are undoubtedly one of the most common and important 3-atom components and have been widely used in 1,3-dipolar cycloaddition for the construction of optically active heterocycles.1a However, the research interest has been prominently focused on the [3+2] and [3+3] cycloadditions,42 and only a few works on the investigation of the [3+4] cycloaddition between 1,3-dipoles and dienes have been reported, probably ascribed to the tendency for reactions between 1,3-dipoles and dienes to favour the [3+2] pathway rather than the [3+4] pathway.43 Considering that the azoalkene has proven to be a promising 4-atom participant, we sought to evaluate the feasibility of enantioselective 1,3-dipolar [3+4] cycloaddition between the 1,3-dipoles and azoalkenes.Nitrone was selected as the representative 1,3-dipole because of its availability and stability. It was found that the [3+4] cycloadduct could be obtained in good yield either with or without catalysts. Obviously, the strong background reaction restricts the development of the asymmetric variant of this annulation. The condition optimization indicated that the N-protecting groups of azoalkenes, the N-substituents of nitrones, the solvents and the reaction temperature all have a noticeable impact on the enantioselectivity. α-Chloro N-acyl hydrazones and N-Me nitrones 47 are superior reaction partners catalysed by the CuBF4/iPr-phosferrox(L4) complex, giving the corresponding [3+4] cycloadduct 48 in high yields with good to excellent enantioselectivities. However, some limitations were observed with this protocol. For alkyl-substituted nitrones or azoalkene precursors, only racemic products were obtained, and no reaction occurred with ortho-substituted hydrazone (Scheme 21).44
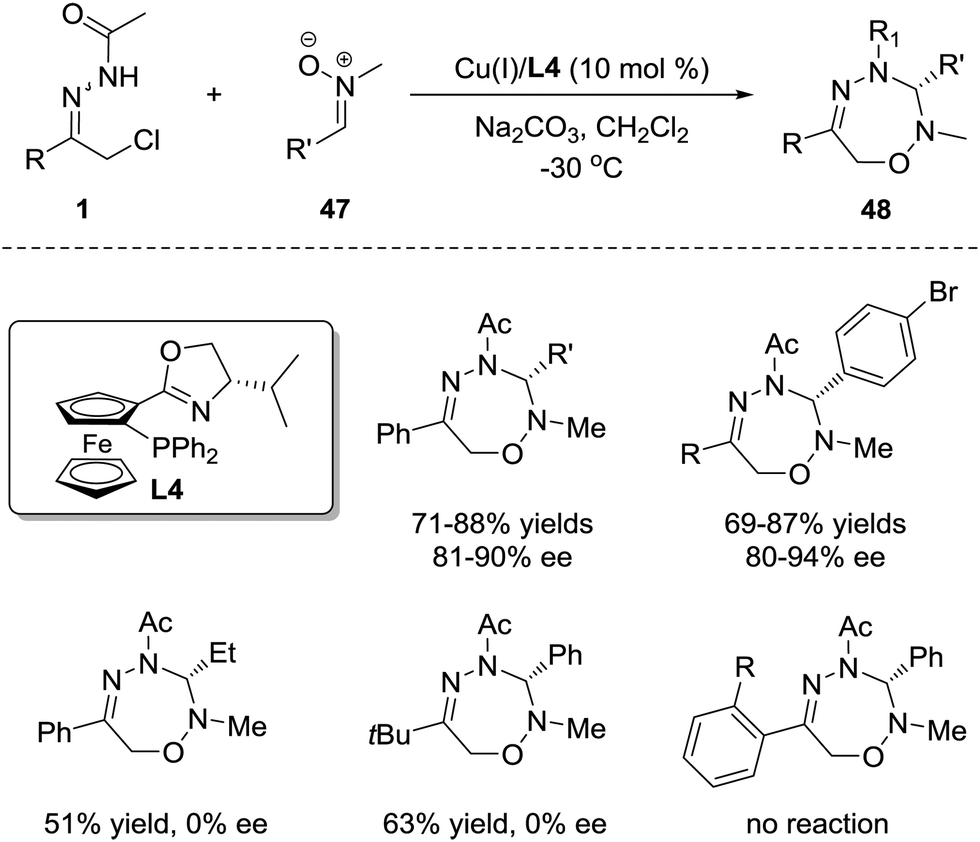 | ||
| Scheme 21 Cu(I)-Catalyzed asymmetric 1,3-dipolar [4+3] cycloaddition of azoalkenes with nitrones and its limitations. | ||
Zhao et al. simultaneously reported a racemic version of a similar [3+4] cycloaddition of nitrones with azoalkenes. A diverse set of 1,2,4,5-oxatriazepane derivatives were synthesized without any catalysts, demonstrating that the reaction between azoalkenes and 1,3-dipoles is a practical and efficient route to seven-membered heterocycles.45
Recently, we demonstrated that the 1,3-dipolar [3+4] cycloaddition of azoalkene can be extended to other stable 1,3-dipoles such as azomethine imine 49.46 This transformation shows better stereoselectivity and broader substrate scope towards both 1,3-dipoles and azoalkenes compared with the reaction between nitrones and azoalkenes. Furthermore, a kinetic resolution of racemic azomethine imines could be realized using the same protocol with high S factors (Scheme 22).
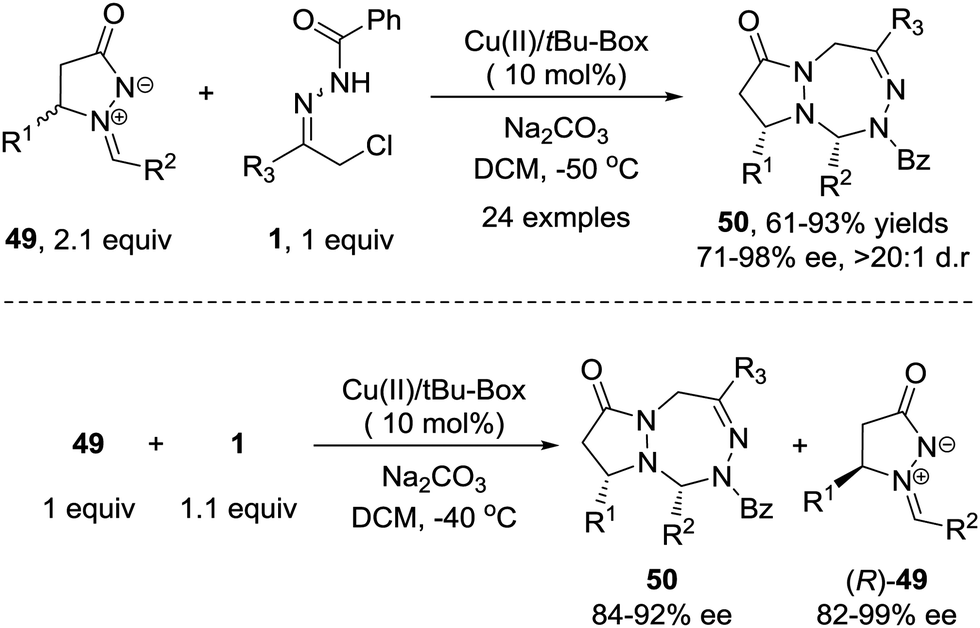 | ||
| Scheme 22 Cu(II)-Catalyzed asymmetric 1,3-dipolar [4+3] cycloaddition and kinetic resolution of azoalkenes with azomethine imines. | ||
Other types of reactions of azoalkene
Azoalkenes are not only suitable 4-atom components but also can behave as electrophiles in conjugated addition to react with various nucleophiles.In 2010, the Coltart group reported a Cu(I)-catalysed addition of Grignard reagents to in situ-derived N-sulfonyl azoalkenes 51.47 At least 2 equivalents of the Grignard reagent are needed in this reaction because the Grignard reagent both acts as a base to produce azoalkene and undergoes transmetalation with the Cu(I) catalyst to form a cuprate capable of conjugate addition to azoalkene. Due to the highly reactive nature of both azoalkenes and Grignard reagents, this umpolung alkylation strategy enables the synthesis of extremely hindered compounds that would be inaccessible using traditional enolate chemistry. Starting from N-sulfonyl hydrazones, the sequential oxidation/alkylation process is also feasible. Additionally, one-pot α,α-bisalkylations have been successfully developed using α,α-dichloro-N-sulfonyl hydrazones 53 as the starting materials (Scheme 23).
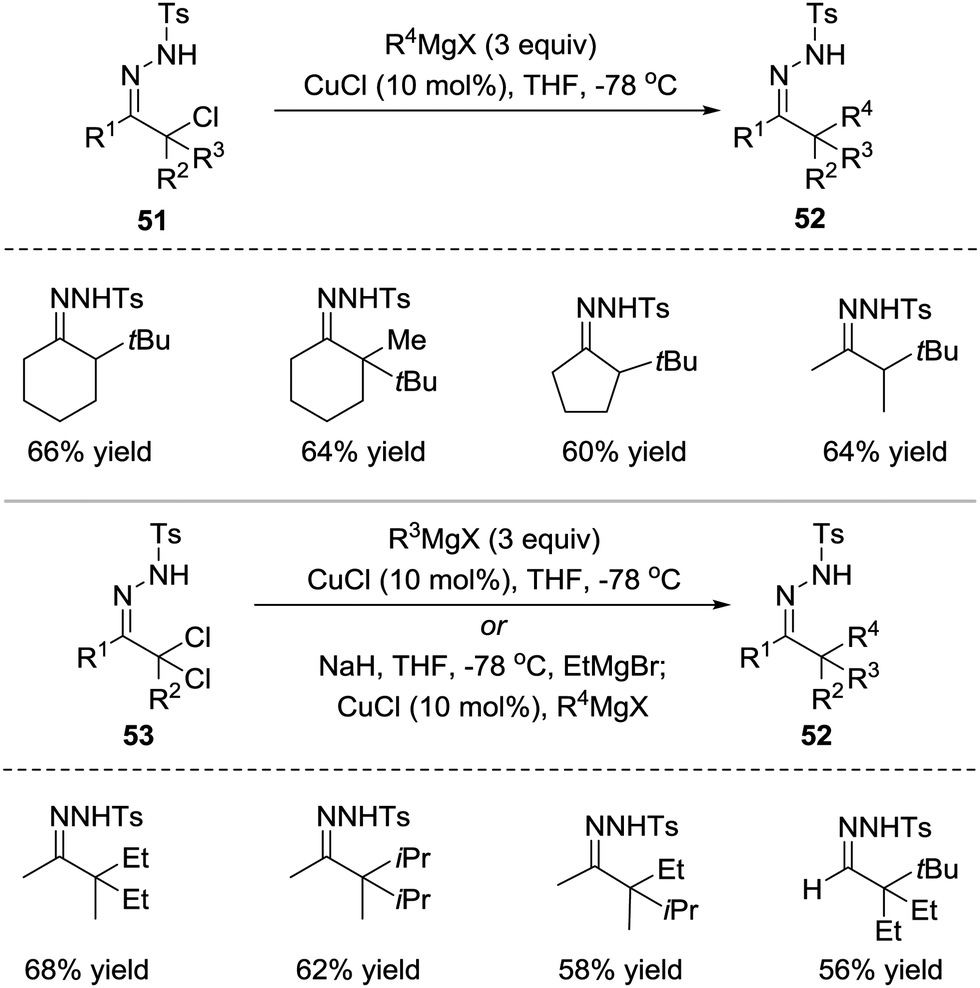 | ||
| Scheme 23 Cu(I)-Catalyzed conjugate addition of Grignard reagents to azoalkenes. | ||
Later, in 2015, the Coltart group reported the conjugate addition of Grignard reagents with α-epoxy N-sulfonyl hydrazones 54.48 Unlike the α-halo hydrazones, the deprotonation of α-epoxy N-sulfonyl hydrazone by Grignard reagents triggers ring opening of the epoxide and leads to the 3-alkoxy-1-N-sulfonyl azopropene intermediate. The alkoxide of the intermediate would then coordinate to the magnesium of the Grignard reagent, which plays a crucial role in this transformation for both reactivity and selectivity. The authors believe the coordination not only induces addition but also causes it to proceed in the desired 1,4-manner. Furthermore, the coordination forces the Grignard reagents to attack from the same face of the azoalkene, thus resulting in a highly diastereoselective addition. Remarkably, this transformation is able to incorporate an unprecedented scope of carbon-based substituents, including 1°, 2°, and 3° alkyls, as well as the alkenyl, aryl, allenyl and alkynyl groups (Scheme 24).
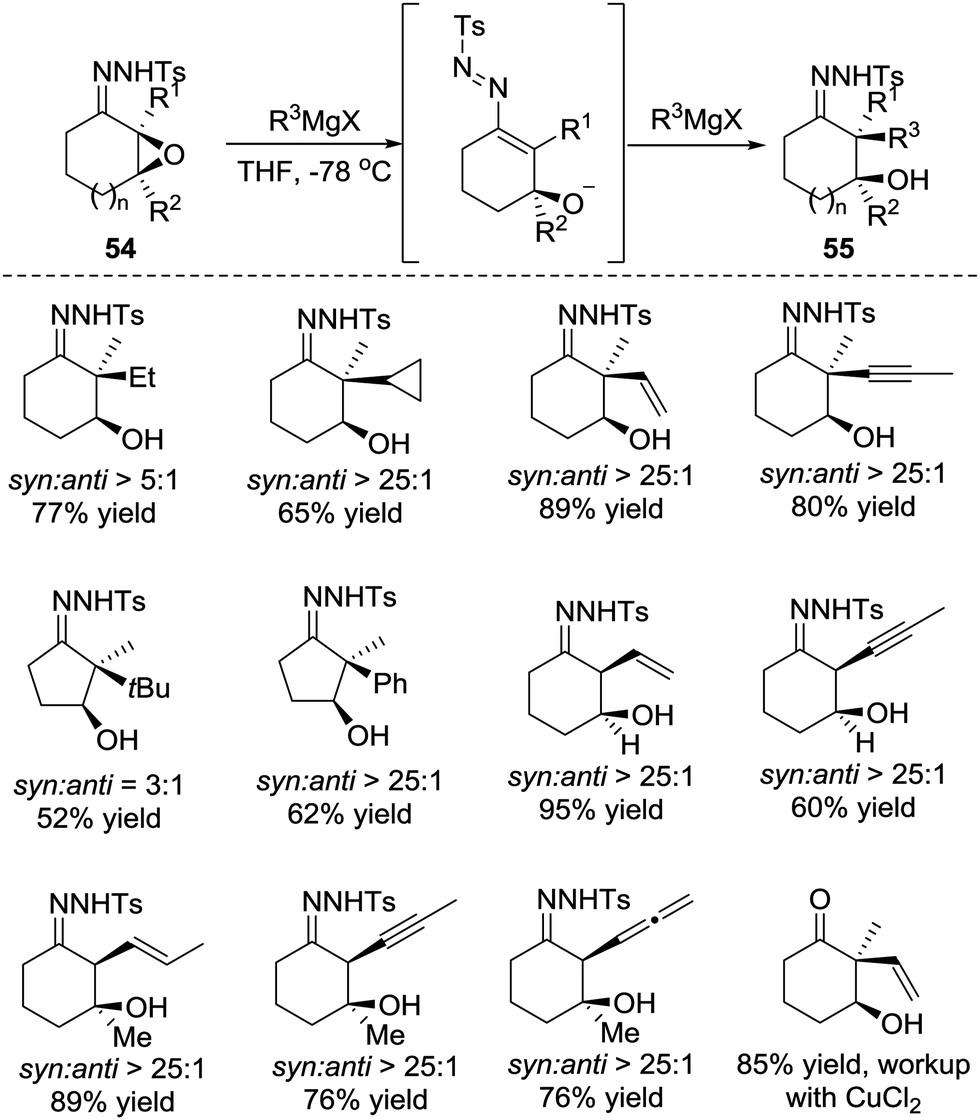 | ||
| Scheme 24 Diastereoselective addition of Grignard reagents to α-epoxy N-sulfonyl hydrazones. | ||
In the same year, Toste et al. realized a chiral phosphoric acid 58 catalysed conjugate addition of anilines to azoalkenes 56, which provide a novel strategy for the enantioselective synthesis of α-amino ketones.49 Unlike other reports that azoalkenes are usually activated by Lewis acids, this transformation demonstrated that the interaction between azoalkenes and Brønsted phosphoric acid50 under the appropriate conditions will also result in the formation of an activated intermediate HA*-56 (Scheme 25).
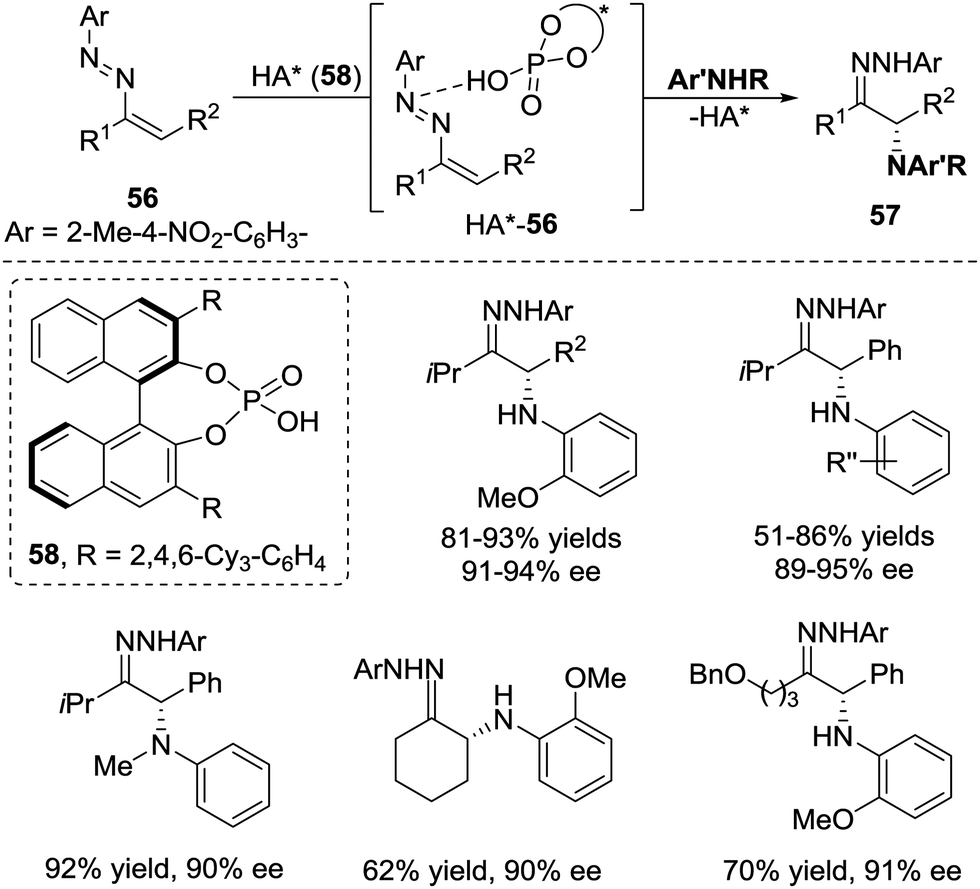 | ||
| Scheme 25 Chiral phosphoric acid catalyzed α-amination of azoalkenes. | ||
A cascade vinylogous 1,6-Michael addition/1,4-proton shift/aza-Michael addition/hemiaminal cyclization process of activated azoalkenes 59 with β-substituted 2-butenals 60 was developed by the Chen group in 2017, which provided a directed entry to a variety of 8-diazabicyclo[3.3.0]-octane derivatives 63 bearing a quaternary stereogenic centre in 43–95% yields, >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r. with 31 ≥ 99% ee (Scheme 26).51
:1 d.r. with 31 ≥ 99% ee (Scheme 26).51
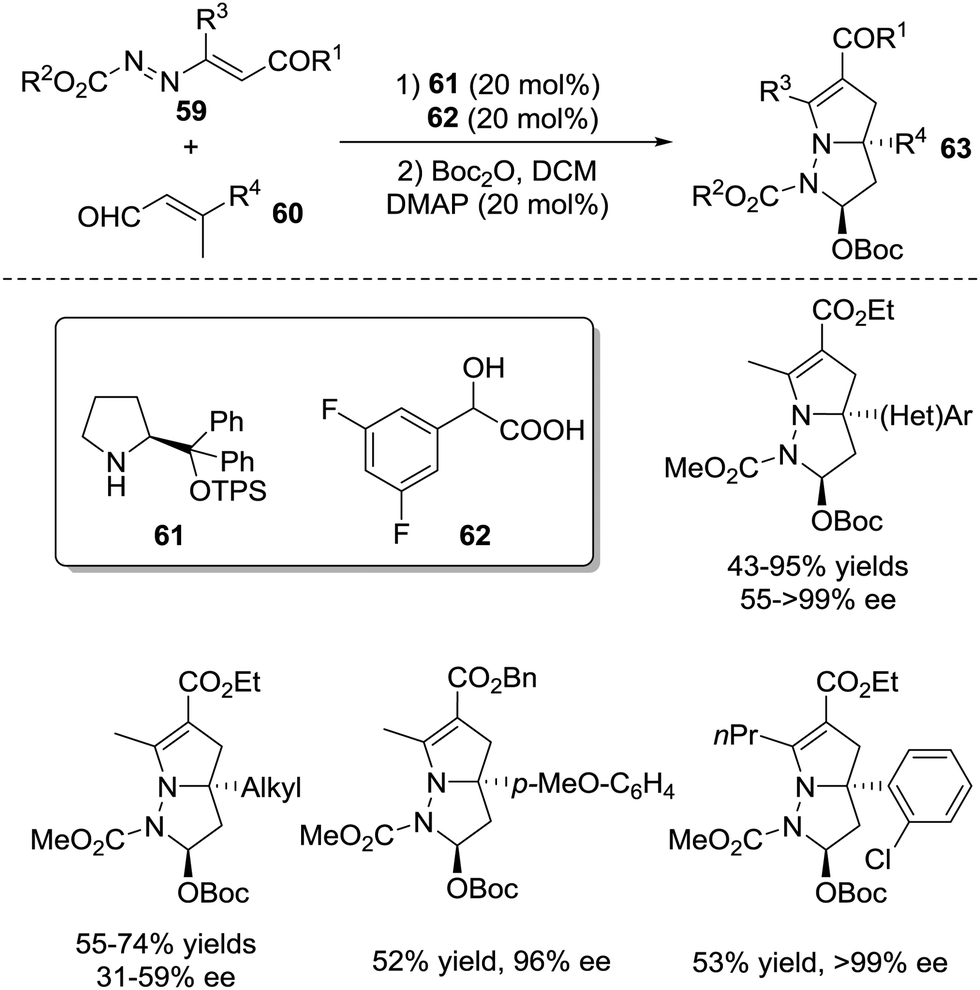 | ||
| Scheme 26 Amine/acid catalyzed asymmetric cascade reaction of azoalkenes. | ||
The authors proposed a possible reaction mechanism to explain the unprecedented cascade process. As shown in Scheme 27, the dienamine intermediate I generated from amine 61 and enal 60 attacked activated azoalkene 59 in a remote 1,6-vinylogous Michael addition manner, giving intermediate II, which then transformed to the N-anion intermediate IIIvia tautomerization. Subsequently, a key 1,4-proton shift occurred to produce intermediate IV, followed by an enantiospecific intramolecular aza-Michael addition from the Re-face, delivering intermediate V. The hydrolysis of V would regenerate amine 61 and afford aldehyde VI. The spontaneous intramolecular hemiaminalization of VI would produce the observed bicyclic compound 63.
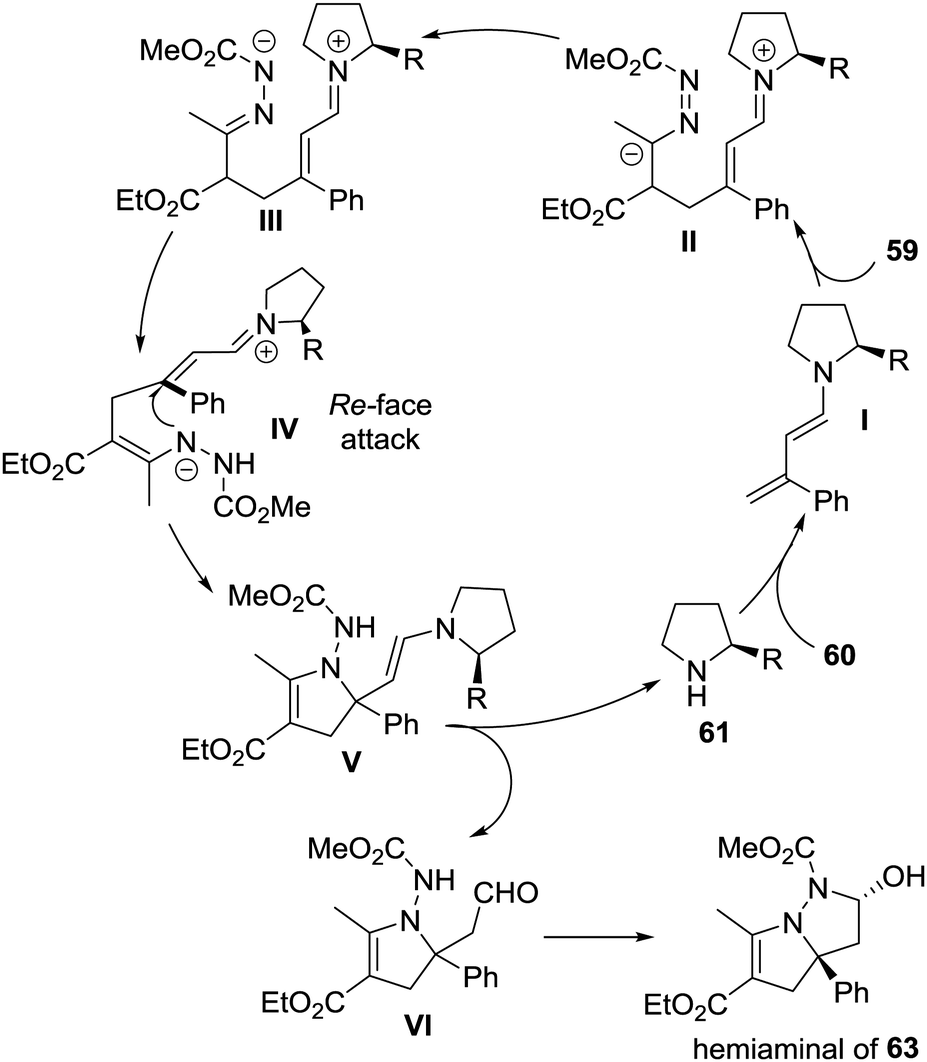 | ||
| Scheme 27 Proposed mechanism for the cascade reaction of activated azoalkene with α-substituted 2-butenals. | ||
Conclusions
Catalytic asymmetric annulations of azoalkenes have become a valid tool to construct optically active multi-nitrogen-containing heterocycles. In particular, the highly reactive nature of azoalkene allows the reaction to proceed under mild conditions with many types of reaction partners, including sulfur ylides, diazo esters, aldehydes, anhydrides, olefins and 1,3-dipoles. With these methodologies, heterocycles with diverse ring sizes and structures can be constructed in high yields with high levels of stereocontrol obtained with an asymmetric catalysis strategy. Further applications of azoalkenes are worth pursuing with new reaction development for the synthesis of other heterocycles, biologically active compounds and natural products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (21525207, 21772147), China Postdoctoral Science Foundation funded project (2017M620331), and the Fundamental Research Funds for the Central Universities. The Program of Introducing Talents of Discipline to Universities of China (111 Program) is also appreciated. We also sincerely thank all co-workers who have contributed to the work described in this account.Notes and references
- (a) Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, ed. A. Padwa and W. H. Pearson, Wiley, 2003 Search PubMed; (b) A. Bianco, M. Maggini, G. Scorrano, C. Toniolo, G. Marconi, C. Villani and M. Prato, J. Am. Chem. Soc., 1996, 118, 4072 CrossRef CAS; (c) F.-X. Felpin and J. Lebreton, Eur. J. Org. Chem., 2003, 3693 CrossRef CAS; (d) Y. Chen, Z.-T. Huang and M.-X. Wang, Curr. Org. Chem., 2004, 8, 325–351 CrossRef; (e) J. P. Michael, Nat. Prod. Rep., 2005, 22, 603–626 RSC.
- (a) W. H. Pearson, Pure Appl. Chem., 2002, 74, 1339 CAS; (b) W. H. Pearson and P. Stoy, Synlett, 2003, 0903 CrossRef CAS; (c) X.-X. Jiang and R. Wang, Chem. Rev., 2013, 113, 5515 CrossRef CAS PubMed; (d) K. A. Jørgensen, Angew. Chem., Int. Ed., 2000, 39, 3558 CrossRef; (e) K. A. Jørgensen, Cycloaddition reactions in organic synthesis, John Wiley & Sons, Chicester, 2002 Search PubMed.
- (a) F. Bellina and R. Rossi, Curr. Org. Chem., 2004, 8, 1089 CrossRef CAS; (b) A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199 CrossRef CAS PubMed; (c) H. T. Pham, I. Chataigner and J.-L. Renaud, Curr. Org. Chem., 2012, 16, 1754 CrossRef CAS; (d) A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2014, 43, 5750 RSC; (e) W. Cieslik, M. Serda and R. Musiol, Curr. Org. Chem., 2015, 19, 1410 CrossRef CAS; (f) H. Ni, G. Zhang and Y. Yu, Curr. Org. Chem., 2015, 19, 776–789 CrossRef CAS.
- (a) O. A. Attanasi, L. De Crescentini, P. Filippone, F. Mantellini and S. Santeusanio, ARKIVOC, 2002, 11, 274 Search PubMed; (b) O. A. Attanasi, L. De Crescentini, G. Favi, P. Filippone, F. Mantellini, F. R. Perrulli and S. Santeusanio, Eur. J. Org. Chem., 2009, 3109 CrossRef CAS.
- L. Wei, Y. Zhou, Z.-M. Song, H.-Y. Yao, Z. Lin and C.-J. Wang, Chem. – Eur. J., 2017, 23, 4995 CrossRef CAS PubMed.
- (a) C.-H. Küchenthal and W. Maison, Synthesis, 2010, 719 Search PubMed; (b) M. Kissane and A. R. Maguire, Chem. Soc. Rev., 2010, 39, 845–883 RSC; (c) L. Albert, J. Heterocycl. Chem., 2002, 39, 1–13 CrossRef.
- (a) P. Dalla Croce, Ann. Chim., 1973, 63, 895 CAS; (b) J.-R. Chen, W.-R. Dong, M. Candy, F.-F. Pan, M. Jörres and C. Bolm, J. Am. Chem. Soc., 2012, 134, 6924–6927 CrossRef CAS PubMed.
- (a) Z. Wang, Y. Yang, F. Gao, Z. Wang, Q. Luo and L. Fang, Org. Lett., 2018, 20, 934 CrossRef CAS PubMed; (b) J. Shao, W. Chen, M. Zhao, K. Shu, H. Liu and P. Tang, Org. Lett., 2018, 20, 3996 CrossRef PubMed.
- O. A. Attanasi, L. De Crescentini, G. Favi, F. Mantellini, S. Mantenuto and S. Nicolini, J. Org. Chem., 2014, 79, 8331 CrossRef CAS PubMed.
- R. Huang, H.-Y. Tao and C.-J. Wang, Org. Lett., 2017, 19, 1176–1179 CrossRef CAS PubMed.
- (a) M. Yang, T. Wang, S. Cao and Z. He, Chem. Commun., 2014, 50, 13506 RSC; (b) X.-N. Zhang, G.-Q. Cheng, X.-Y. Tang, Y. Wei and M. Shi, Angew. Chem., Int. Ed., 2014, 53, 10768–10773 CrossRef CAS PubMed.
- (a) D. W. Combs, K. Reese, L. A. M. Cornelius, J. W. Gunnet, E. V. Cryan, K. S. Granger, J. J. Jordan and K. T. Demarest, J. Med. Chem., 1995, 38, 4880 CrossRef CAS PubMed; (b) D. W. Combs, K. Reese and A. Phillips, J. Med. Chem., 1995, 38, 4878 CrossRef CAS PubMed; (c) Z. Lijun, M. A. Williams, D. B. Mendel, P. A. Escarpe, C. Xiaowu, K.-Y. Wang, J. Graves, B. G. Lawton and C. U. Kim, Bioorg. Med. Chem. Lett., 1999, 9, 1751 CrossRef; (d) J. Y. Cho, H. C. Kwon, P. G. Williams, P. R. Jensen and W. Fenical, Org. Lett., 2006, 8, 2471 CrossRef CAS PubMed; (e) J. H. M. Lange, A. P. Den Hartog, M. a. W. Van Der Neut, B. J. Van Vliet and C. G. Kruse, Bioorg. Med. Chem. Lett., 2009, 19, 5675 CrossRef CAS PubMed.
- (a) M. S. South, T. L. Jakuboski, M. D. Westmeyer and D. R. Dukesherer, J. Org. Chem., 1996, 61, 8921 CrossRef CAS PubMed; (b) M. D’Auria, R. Racioppi, O. A. Attanasi and F. Mantellini, Synlett, 2010, 1363 CrossRef; (c) S. M. M. Lopes, A. F. Brigas, F. Palacios, A. Lemos and T. M. V. D. Pinho e Melo, Eur. J. Org. Chem., 2012, 2152 CrossRef CAS; (d) F. Palacios, D. Aparicio, Y. López, J. M. de los Santos and C. Alonso, Eur. J. Org. Chem., 2005, 1142–1147 CrossRef CAS.
- G. Pitacco, O. A. Attanasi, L. D. Crescentini, G. Favi, F. Felluga, C. Forzato, F. Mantellini, P. Nitti, E. Valentin and E. Zangrando, Tetrahedron: Asymmetry, 2010, 21, 617–622 CrossRef CAS.
- X. Li, K. Gai, Z. Yuan, J. Wu, A. Li and H. Yao, Adv. Synth. Catal., 2015, 357, 3479 CrossRef CAS.
- S. Gao, J.-R. Chen, X.-Q. Hu, H.-G. Cheng, L.-Q. Lu and W.-J. Xiao, Adv. Synth. Catal., 2013, 355, 3539 CrossRef CAS.
- L. Wei and C.-J. Wang, Chem. Commun., 2015, 51, 15374 RSC.
- (a) P. M. Dewick, Medicinal Natural Products: A Biosynthetic Approach, Wiley, New York, 2nd edn, 2002 Search PubMed; (b) E. Fattorusso and O. T. Scafati, Modern Alkaloids, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) J. F. Austin, S.-G. Kim, C. J. Sinz, W.-J. Xiao and D. W. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5482 CrossRef CAS PubMed; (b) Y.-C. Xiao, C. Wang, Y. Yao, J. Sun and Y.-C. Chen, Angew. Chem., Int. Ed., 2011, 50, 10661 CrossRef CAS PubMed; (c) Q. Cai, C. Zheng, J.-W. Zhang and S.-L. You, Angew. Chem., Int. Ed., 2011, 50, 8665 CrossRef CAS PubMed; (d) B. M. Trost and J. Quancard, J. Am. Chem. Soc., 2006, 128, 6314 CrossRef CAS PubMed; (e) O. Lozano, G. Blessley, T. Martinez del Campo, A. L. Thompson, G. T. Giuffredi, M. Bettati, M. Walker, R. Borman and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 8105 CrossRef CAS PubMed; (f) B. Tan, G. Hernández-Torres and C. F. Barbas, J. Am. Chem. Soc., 2011, 133, 12354 CrossRef CAS PubMed; (g) G. Cera, M. Chiarucci, A. Mazzanti, M. Mancinelli and M. Bandini, Org. Lett., 2012, 14, 1350 CrossRef CAS PubMed; (h) J. Barluenga, E. Tudela, A. Ballesteros and M. Tomás, J. Am. Chem. Soc., 2009, 131, 2096 CrossRef CAS PubMed; (i) Y. Lian and H. M. L. Davies, J. Am. Chem. Soc., 2010, 132, 440 CrossRef CAS PubMed; (j) L. M. Repka, J. Ni and S. E. Reisman, J. Am. Chem. Soc., 2010, 132, 14418 CrossRef CAS PubMed; (k) Q. Cai and S.-L. You, Org. Lett., 2012, 14, 3040 CrossRef CAS PubMed; (l) Z. Chen, B. Wang, Z. Wang, G. Zhu and J. Sun, Angew. Chem., Int. Ed., 2013, 52, 2027 CrossRef CAS PubMed; (m) H. Xiong, H. Xu, S. Liao, Z. Xie and Y. Tang, J. Am. Chem. Soc., 2013, 135, 7851 CrossRef CAS PubMed.
- S. L. Schreiber, Science, 2000, 287, 1964 CrossRef CAS PubMed.
- M.-C. Tong, X. Chen, J. Li, R. Huang, H. Tao and C.-J. Wang, Angew. Chem., Int. Ed., 2014, 53, 4680 CrossRef CAS PubMed.
- (a) G. Casiraghi, F. Zanardi, G. Appendino and G. Rassu, Chem. Rev., 2000, 100, 1929 CrossRef CAS PubMed; (b) S. F. Martin, Acc. Chem. Res., 2002, 35, 895 CrossRef CAS PubMed; (c) S. E. Denmark, J. R. Heemstra Jr. and G. L. Beutner, Angew. Chem., Int. Ed., 2005, 44, 4682 CrossRef CAS PubMed; (d) G. Casiraghi, L. Battistini, C. Curti, G. Rassu and F. Zanardi, Chem. Rev., 2011, 111, 3076 CrossRef CAS PubMed; (e) R. C. FusoN, Chem. Rev., 1935, 16, 1 CrossRef CAS.
- (a) B. M. Fraga, Nat. Prod. Rep., 2002, 19, 650 RSC; (b) S.-X. Huang, L.-B. Yang, W.-L. Xiao, C. Lei, J.-P. Liu, Y. Lu, Z.-Y. Weng, L.-M. Li, R.-T. Li, J.-L. Yu, Q.-T. Zheng and H.-D. Sun, Chem. – Eur. J., 2007, 13, 4816 CrossRef CAS PubMed; (c) H. N. Kamel, D. Ferreira, L. F. Garcia-Fernandez and M. Slattery, J. Nat. Prod., 2007, 70, 1223 CrossRef CAS PubMed; (d) J. S. Roach, P. LeBlanc, N. I. Lewis, R. Munday, M. A. Quilliam and S. L. MacKinnon, J. Nat. Prod., 2009, 72, 1237 CrossRef CAS PubMed.
- J. Li, R. Huang, Y.-K. Xing, G. Qiu, H.-Y. Tao and C.-J. Wang, J. Am. Chem. Soc., 2015, 137, 10124 CrossRef CAS PubMed.
- E. M. Vogl, H. Gröger and M. Shibasaki, Angew. Chem., Int. Ed., 1999, 38, 1570 CrossRef CAS PubMed.
- D. A. Singleton and A. A. Thomas, J. Am. Chem. Soc., 1995, 117, 9357 CrossRef CAS.
- (a) M. Harmata, Acc. Chem. Res., 2001, 34, 595 CrossRef CAS PubMed; (b) M. Harmata, Chem. Commun., 2010, 46, 8904 RSC; (c) A. G. Lohse and R. P. Hsung, Chem. – Eur. J., 2011, 17, 3812 CrossRef CAS PubMed.
- R. Huang, X. Chang, J. Li and C.-J. Wang, J. Am. Chem. Soc., 2016, 138, 3998 CrossRef CAS PubMed.
- X. Xu and M. P. Doyle, Acc. Chem. Res., 2014, 47, 1396 CrossRef CAS PubMed.
- Y. Deng, C. Pei, H. Arman, K. Dong, X. Xu and M. P. Doyle, Org. Lett., 2016, 18, 5884 CrossRef CAS PubMed.
- X. Zhong, J. Lv and S. Luo, Org. Lett., 2015, 17, 1561 CrossRef CAS PubMed.
- (a) I. Fleming, A. Barbero and D. Walter, Chem. Rev., 1997, 97, 2063 CrossRef CAS PubMed; (b) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388 CrossRef CAS PubMed.
- R. Hoffmann and R. B. Woodward, J. Am. Chem. Soc., 1965, 87, 4388 CrossRef CAS.
- (a) E. D. Bergmann, Chem. Rev., 1968, 68, 41 CrossRef CAS; (b) P. Preethalayam, K. S. Krishnan, S. Thulasi, S. S. Chand, J. Joseph, V. Nair, F. Jaroschik and K. V. Radhakrishnan, Chem. Rev., 2017, 117, 3930 CrossRef CAS PubMed.
- L. Wei, Q. Zhu, Z.-M. Song, K. Liu and C.-J. Wang, Chem. Commun., 2018, 54, 2506 RSC.
- I. V. Hartung and H. M. R. Hoffmann, Angew. Chem., Int. Ed., 2004, 43, 1934 CrossRef CAS PubMed.
- (a) T. V. Nguyen, J. M. Hartmann and D. Enders, Synthesis, 2013, 845 CAS; (b) M. A. Battiste, P. M. Pelphrey and D. L. Wright, Chem. – Eur. J., 2006, 12, 3438 CrossRef CAS PubMed; (c) H. Butenschön, Angew. Chem., Int. Ed., 2008, 47, 5287 CrossRef PubMed; (d) K. E. O. Ylijoki and J. M. Stryker, Chem. Rev., 2013, 113, 2244 CrossRef CAS PubMed.
- (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed; (b) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011, 44, 1182 CrossRef CAS PubMed; (c) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC; (d) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307 CrossRef CAS PubMed.
- C. Guo, B. Sahoo, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17402 CrossRef CAS PubMed.
- J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2014, 47, 696 CrossRef CAS PubMed.
- L. Wang, S. Li, M. Blümel, A. R. Philipps, A. Wang, R. Puttreddy, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2016, 55, 11110 CrossRef CAS PubMed.
- K. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366 CrossRef PubMed.
- (a) B. M. Trost and D. T. MacPherson, J. Am. Chem. Soc., 1987, 109, 3483 CrossRef CAS; (b) R. Shintani, M. Murakami and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 12356 CrossRef CAS PubMed.
- L. Wei, L. Yao, Z.-F. Wang, H. Li, H.-Y. Tao and C.-J. Wang, Adv. Synth. Catal., 2016, 358, 3748 CrossRef CAS.
- (a) X.-Q. Hu, J.-R. Chen, S. Gao, B. Feng, L.-Q. Lu and W.-J. Xiao, Chem. Commun., 2013, 49, 7905 RSC; (b) H.-W. Zhao, H.-L. Pang, T. Tian, B. Li, X.-Q. Chen, X.-Q. Song, M. Wei, Z. Yang, Y.-Y. Liu and Y.-D. Zhao, Adv. Synth. Catal., 2016, 358, 1826 CrossRef CAS.
- L. Wei, Z.-F. Wang, L. Yao, G. Qiu, H. Tao, H. Li and C.-J. Wang, Adv. Synth. Catal., 2016, 358, 3955 CrossRef CAS.
- J. M. Hatcher and D. M. Coltart, J. Am. Chem. Soc., 2010, 132, 4546 CrossRef CAS PubMed.
- M. M. Uteuliyev, T. T. Nguyen and D. M. Coltart, Nat. Chem., 2015, 7, 1024 CrossRef CAS PubMed.
- D. H. Miles, J. Guasch and F. D. Toste, J. Am. Chem. Soc., 2015, 137, 7632 CrossRef CAS PubMed.
- (a) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed; (b) R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603 CrossRef CAS PubMed.
- G.-Y. Ran, M. Gong, J.-F. Yue, X.-X. Yang, S.-L. Zhou, W. Du and Y.-C. Chen, Org. Lett., 2017, 19, 1874 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |