
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rhenium is different: CO tetramerization induced by a divalent lanthanide complex in rhenium carbonyls†
Ravi
Yadav
,
Thomas
Simler
 ,
Michael T.
Gamer
,
Ralf
Köppe
and
Peter W.
Roesky
*
,
Michael T.
Gamer
,
Ralf
Köppe
and
Peter W.
Roesky
*
Institute of Inorganic Chemistry, Karlsruhe Institute of Technology, Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: roesky@kit.edu
First published on 30th April 2019
Abstract
The reduction of M2(CO)10 (M = Mn, Re) with different divalent lanthanide (Ln = Sm, Yb) compounds was investigated. Depending on the steric demand of the ligand, either unusual CO tetramerization or formation of a new Re carbonyl anion occurred in the case of Re. Theoretical calculations were performed for a better understanding of the nature of bonding in the newly formed species.
The coupling of carbon monoxide in the presence of hydrogen to form short hydrocarbon chains is performed on an industrial scale in the Fischer–Tropsch process. Thus, carbon monoxide is a key C1 feedstock for the industrial production of hydrocarbons. The primary products of the Fischer–Tropsch synthesis are alkanes, alkenes, and to a minor extent alcohols, which are all valuable starting materials in the chemical industry.1 The product selectivity in the Fischer–Tropsch process can be varied over a wide range. Key parameters are the catalyst formulation and the reaction conditions. The chain growth probability is influenced by the catalyst that is selected for the hydrogenation process.2 Industrially, mostly Fe or Co catalysts have been employed in this reaction.3 For a better understanding of the Fischer–Tropsch process, numerous metal complexes, which promote the coupling of CO have been published.4 For example, CO dimerization has been observed using Th–H,4d Zr–H,4e Ce–H,4f or Mg–H/Ca–H4g,h catalysts. However, the number of complexes forming higher coupling products (C3, C4, and C6) is very limited. Examples have been published using actinides (C3O32−,5 C4O44−,5b,6 OC(CH2-tBu)C(O)
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(O)–C(CH2-tBu)O2−),7 lanthanides {(O2C–CCO)2−,8 OC(tBu)C(O)C(O)–C(tBu)O2−,9 and a [BC(O)C(O)C(O)B] skeleton10}, tantalum (C6O68−),11 iron (C4(OR)4),12 and very recently [W(CO)6] and Al(I) ([C3O3]4− and [C4O4]4−).4c
C(O)–C(CH2-tBu)O2−),7 lanthanides {(O2C–CCO)2−,8 OC(tBu)C(O)C(O)–C(tBu)O2−,9 and a [BC(O)C(O)C(O)B] skeleton10}, tantalum (C6O68−),11 iron (C4(OR)4),12 and very recently [W(CO)6] and Al(I) ([C3O3]4− and [C4O4]4−).4c
The reductive behaviour of 5d transition-metal (TM) carbonyl complexes is far less explored as compared with their lighter analogues. In 2017, Deacon and co-workers trapped the elusive [W2(CO)10]2− anion featuring an unsupported W–W bond by the reduction of W(CO)6 with a divalent samarium meso-octaethylcalix[4]pyrrolide.13 Recently, single-electron reduced rhenium carbonyl complexes have shown electrocatalytic reduction of CO2 to CO.14
Herein, we report the reduction of Re2(CO)10 with divalent lanthanide complexes, which leads to a tetramerization of CO to give a Fischer-type rhenacycle. We also studied the effect of the ligands on the reduction ability of the lanthanide complexes and compared the reactivity of Re2(CO)10 to that of Mn2(CO)10.
Reaction of [(Cp*)2SmII(thf)2]15 (Cp* = C5Me5) with Re2(CO)10 in toluene at room temperature furnished [{(Cp*)2Sm}3{(μ-O4C4)(μ-η2-CO)2(μ-η1-CO)(CO)5Re2}Sm(Cp*)2(thf)] (1) as red coloured crystals in 18% yield (Scheme 1). Since SmII is a single-electron reduction reagent,16 the central [(μ-O4C4)(μ-η2-CO)2(μ-η1-CO)(CO)5Re2]4− core has been formed by a four-fold reduction process and the [μ-O4C4] unit can formally be considered as tetra-anionic where each oxygen atom is negatively charged. The solid-state IR spectrum of 1 showed characteristic νCO stretches, which are different from the CO vibrations of Re2(CO)10. Notably, the low-frequency stretches at 1792(s) and 1733(s) cm−1 indicate bridging isocarbonyls. No suitable NMR data could be obtained, most probably due to decomposition in solution. The solid-state structure of 1 revealed the formation of a hexametallic coordination cluster consisting of one [(μ-O4C4)(μ-η2-CO)2(μ-η1-CO)(CO)5Re2]4−, one [(Cp*)2SmIII(thf)]+, and three [(Cp*)2SmIII]+ moieties (Fig. 1). The Sm1, Sm2, and Sm3 atoms are coordinated by two η5-Cp* and two O atoms from bridging isocarbonyls or the newly formed μ-O4C4 entity.
 | ||
| Scheme 1 Synthesis of complex 1 (simplified view) (left) and possible isomers of 1 (right), double Fischer-carbene (1a) and metallacyclopentadiene (1b). | ||
 | ||
| Fig. 1 Molecular structure of 1. Hydrogen atoms are omitted for clarity. Bond lengths and angles are given in ESI.† | ||
The Re–Re bond in 1 is slightly shorter than the Re–Re single bond in Re2(CO)10 (2.934(3) vs. 3.041(11) Å, respectively) but theoretical investigation indicates only a weak interaction.17 The Re1–C1 (2.188(6) Å) and Re1–C4 (2.164(7) Å) bond distances are in the typical range of conjugated rhenacyclic Fischer-type carbenes.18 In principle, two different forms, either a double Fischer-carbene (Scheme 1, 1a) or a metallacyclopentadiene (Scheme 1, 1b), can describe the 5-membered rhenacycle. The short C2–C3 bond distance in the central C4O4 fragment (1.406(9) Å (C2–C3) vs. 1.445(9) Å (C1–C2) and 1.460(9) Å (C3–C4)) suggests the predominance of 1a.18 Thus, we consider compound 1 as a cyclic double Fischer-carbene type complex. Interestingly, formation of the 5-membered Fischer-type rhenacycle in 1 occurred via an unprecedented tetramerization of CO ligands, presumably by reductive C–C coupling. It should be noted that a total of 12 CO units are present in the [(μ-O4C4)(μ-η2-CO)2(μ-η1-CO)(CO)5Re2]4− moiety, more than in the Re2(CO)10 starting material. Since no external source of CO is present, the formation of 1 implies that more than one equivalent of Re2(CO)10 reacts with four equivalents of [(Cp*)2SmII(thf)2]. To the best of our knowledge, CO tetramerization has never been observed with rhenium carbonyls. Albeit two reports describe a similar tetramerization of CO ligands, both involve reactions between trimethylsilylhalide (halide = Br, or I) or [{(Me3Si)2N}BBr2] and [Na2Fe(CO)4].12 Furthermore, the resulting products did not feature Fischer-carbene type character but metallacyclopentadiene type character (Scheme 1, 1b). The reactivity observed is in sharp contrast to all the reports on Ln–TM carbonyl complexes discussed earlier.13,19
Recently, we have shown that using sterically demanding ligands in the coordination sphere of divalent lanthanides, different activations of main-group elements and complexes can be achieved.20 We therefore investigated the reduction of TM carbonyls with [(DippForm)2SmII(thf)2]21 (DippForm = N,N′-bis(2,6-diisopropylphenyl)formamidinate), another sterically demanding divalent samarium reagent. Reaction with half of an equivalent of Re2(CO)10 resulted in the formation of [{(DippForm)2SmIII(thf)}2{(μ-η2-CO)2(μ-η1-CO)2(CO)4Re2}] (2) (Scheme 2) as red coloured crystals in 62% yield. The solid-state IR spectrum of 2 showed characteristic νCO stretches. The low-frequency stretch at 1804 cm−1 is consistent with the occurrence of bridging isocarbonyls.14a The solid-state structure (Fig. 2) confirmed the identity of 2, which is formed through the transfer of 2e− from two molecules of [(DippForm)2SmII(thf)2] to one molecule of Re2(CO)10 in single-electron transfer (SET) steps, accompanied by the loss of two CO ligands. In complex 2, the [Re2(CO)8]2− anion is entrapped between two [(DippForm)2SmIII(thf)]+ moieties. To the best of our knowledge, the [Re2(CO)8]2− anion has never been reported to date. Its lighter analogue, [Mn2(CO)8]2−, has recently been isolated by reduction of Mn2(CO)10 with silylene, however, the authors failed to isolate [Re2(CO)8]2− under similar conditions.22
 | ||
| Scheme 2 Synthesis of complex 2 (simplified view). | ||
 | ||
| Fig. 2 Molecular structure of 2. Hydrogen atoms are omitted for clarity. Bond lengths and angles are given in ESI.† | ||
The isolation of the [(μ-CO)4(CO)4Re2]2− fragment in compound 2 can be traced back to the high reductive nature of divalent samarium and the sterically demanding nature of the [(DippForm)2SmIII(thf)]+ moieties. In the [Re2(CO)8]2− anion, each rhenium atom is coordinated by the carbon donor of two terminal and two bridging CO ligands. The Re–C51 (μ-η2-CO) bond lengths, 2.049(3) and 2.206(3) Å, are longer than the other Re–C bonds due to the η2-type bridging mode with two Re atoms. The Re–Re′ bond length (2.689(3) Å) is substantially shorter than the Re–Re single bond in Re2(CO)10 (3.041(11) Å),17 suggesting a double bond character between the two Re atoms at the first glance.23 Although the [Re2(CO)8]2− anion fits with the 36 electron count only after considering Re–Re double bond, our theoretical calculations suggest a lower bonding order.
The bonding situation in compounds 1 and 2 was investigated by theoretical methods. In many cases the bonding description nicely succeeds by calculation of the shared electron numbers (SEN) according to the population analysis of Ahlrichs and Heinzmann.24 These numbers give a reliable measure of the covalent bond strength, especially even in the case of multi-centre bonding contributions. This effect is referred to as “supported metal–metal bonding” which might appear with the Re–Re contacts in 1 and 2 due to the supporting Re–C–Re bridging. To obtain SEN reference values for an unsupported Re–Re single bond, an analysis of the unbridged Re2(CO)10 molecule was performed. Here, the SEN (Re–Re) is 1.297, but due to a four-centre contribution of the SEN (C–Re–Re–C) = 0.248, about 19% of the Re–Re bond is attributed to this multi-centre bond strengthening. This procedure is justified and was tested by Ahlrichs et al. for the molecule diborane.25 The multi-centre adjusted value would thus be 1.297 − 0.248 = 1.049. As reference for a molecule with a triple-bridged formal Re–Re triple bond, the anion Re2Cl9− was investigated. Here, the SEN is calculated to be 1.915, the three-centre SEN (Re–Cl–Re) is 0.273. Therefore, the pure Re–Re “triple” bond disregarding three-centre bonding effects caused by the three Re–Cl–Re bridges would result in a value of 1.915 − 3 × 0.273 = 1.096.
Applying this method analogously to calculate the covalent bond strengths in compounds 1 and 2, the following conclusions can be drawn: in compound 1, the SEN (Re–Re) = 1.071 and the three-centre contribution is SEN (Re–Re–C) = 0.290, so that for the multi-centre corrected SEN (Re–Re) of 0.491 results. In 2 SEN (Re–Re) = 1.394 and SEN (Re–Re–C) = 0.348, so that SEN (Re–Re) = 0.698, corresponding to a rather weak single bond. The results are summarized in the following way: (i) Re–Re bonding regardless of its origin evaluates in the following row (SEN values in parentheses): 1 (1.071) < Re2(CO)10 (1.297) < 2 (1.394) < Re2Cl9− (1.915). (ii) Taking supporting effects by bridging bonds into account, we conclude that there is virtually no Re–Re bond in 1 and only a weak single two centre Re–Re bond in 2.
To confirm the findings of the population analyses in an orienting manner, we performed a different topological approach by means of the AIM (atoms in molecules) method introduced by Bader.26 The results of the AIM analysis are given in Fig. S10 (ESI†). Compound 2, unlike 1, consists of a bond path with a bond critical point between both rhenium atoms that undoubtedly confirms the presence of a Re–Re bond. The low value of the electron density ρbcp and the high value for the ellipticity ε (ρbcp = 0.05 a.u., ε = 1.31) calculated for this bond critical point of Re–Re indicate a rather weak bond with a high “multiple bond contribution” presumably due to the interaction with the supporting π-type Re–Re–C multicentre bonds.
The bonding properties of the rhenacycle in compound 1 are also interesting. The values for the SEN of the bonds C1–C2, C2–C3, C3–C4 of 1.514, 1.593, 1.518 correspond to strong single bonds. For pure single or double bonds, SEN values of about 1.4 or 2.28,25 respectively, are expected. The SEN (Re–C) is calculated to be 0.95 which is of the same order of magnitude that is also found for the terminal Re–CO bond (1.276). Overall, the results support the view of a double Fischer carbene complex as shown in 1a (Scheme 1).
For comparison, we investigated the reactivity between Mn2(CO)10 and divalent samarium complexes. The reaction of [(Cp*)2Sm(thf)2] with half an equivalent of Mn2(CO)10 in toluene resulted in the formation of [{(Cp*)2Sm(thf)}(μ-CO)2{Mn(CO)3}]n (3) as a red precipitate (Scheme 3). The crystal structure of 3 (Fig. 3) revealed, as expected, cleavage of the Mn–Mn bond upon reduction of Mn2(CO)10 with [(Cp*)2SmII(thf)2] and formation of a [Mn(CO)5]− anion along with [(Cp*)2Sm(thf)]+ as cation. Due to vacant coordination sites on the samarium atom, each [Mn(CO)5]− anion bridges two samarium atoms via isocarbonyl bridges, resulting in a 1D coordination polymer. The average Sm–C (Cp*) bond distance (2.723 Å) is shorter than in [(Cp*)2SmII(thf)2] (2.86(3) Å). The [Mn(CO)5]− anionic fragment has a trigonal bipyramidal geometry with two bridging and three terminal CO. The Mn–C bond lengths of 1.834 Å (terminal, average) and 1.774 Å (bridged, average) are similar to that in [{Cp*2Yb}(μ-CO)3{Mn(CO)2}]n (1.820(5) Å (terminal, average) and 1.791(13) Å (bridged, average)).19d
 | ||
| Scheme 3 Synthesis of complex 3. | ||
 | ||
| Fig. 3 Cutout of the polymeric structure of 3 (left) and molecular structure of 4 (Ln = Sm) and 5 (Ln = Yb) (right). Hydrogen atoms are omitted for clarity. Bond lengths and angles are given in ESI.† | ||
Due to the different reactivity observed with Re2(CO)10 depending on the nature of the ligand on the SmII centre, the reaction between Mn2(CO)10 and [(DippForm)2LnII(thf)2] (Ln = Sm,21 Yb27) was also examined. Half an equivalent of Mn2(CO)10 was reacted with [(DippForm)2LnII(thf)2] (Ln = Sm, Yb) in toluene at 60 °C to give [{(DippForm)2LnIII(thf)}{Mn(CO)5}] (Ln = Sm (4), Yb (5)) (Scheme 4). The low-frequency stretches at 1734 cm−1 (vw) for 4 and 1701 (w) cm−1 for 5 indicate bridging isocarbonyls.19c,d Due to the different steric demand of the Cp* and DippForm ligands, only one [Mn(CO)5]− anion binds to each samarium atom, resulting in discrete molecular species. Complexes 4 and 5 are isostructural and only the structure of 4 is discussed here (see ESI† for 5). In the solid-state structure of 4 (Fig. 3), each Sm atom lies in a distorted octahedral geometry, coordinated by two amidinates, one thf ligand and the oxygen atom of the bridging isocarbonyl. The [Mn(CO)5]− fragment is in a trigonal bipyramidal geometry. Formation of 4 and 5 occurred though cleavage of the Mn–Mn bond via a SET step. The [(DippForm)2SmIII(thf)]+ and [Mn(CO)5]− fragments further assemble by formation of bridging isocarbonyls (Scheme 4).
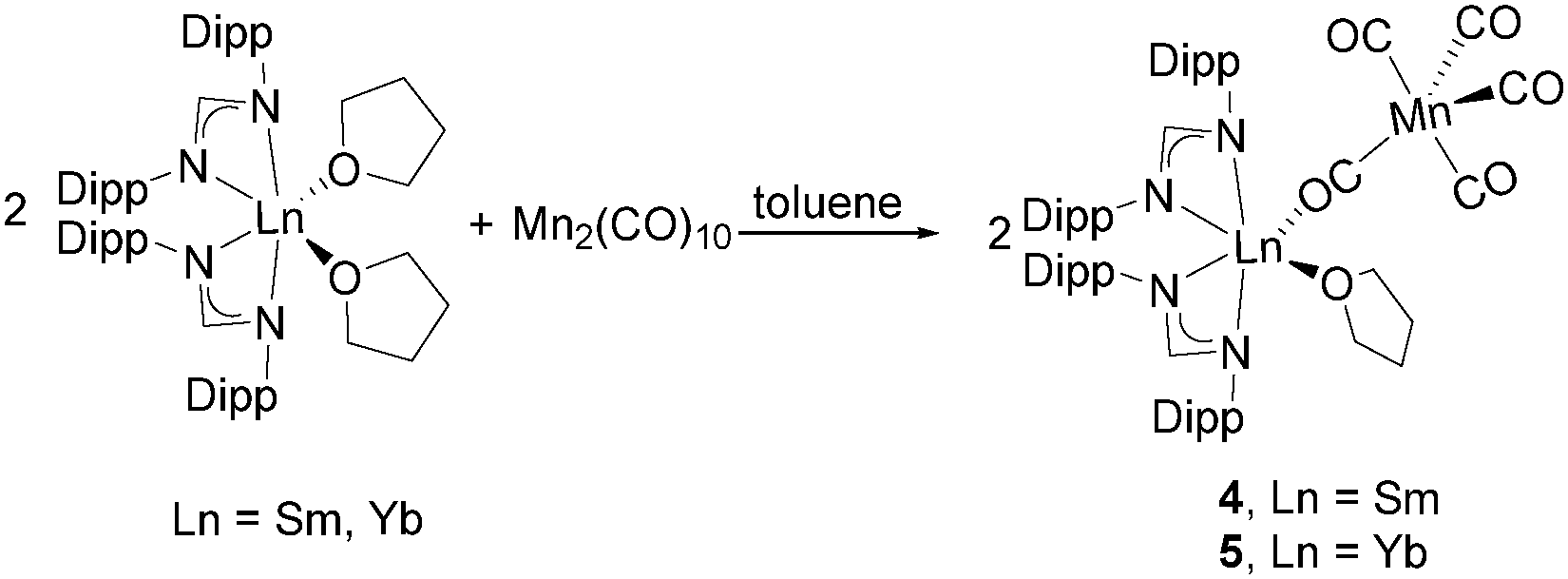 | ||
| Scheme 4 Synthesis of complexes 4 and 5. | ||
In conclusion, we have reacted M2(CO)10 (M = Mn, Re) with different divalent lanthanide compounds. For Mn2(CO)10, the product formation is as expected and the Mn–Mn bond is cleaved upon reduction. The resulting [Mn(CO)5]− anion and the in situ formed [L2SmIII]+ cation (L = Cp*, DippForm) combine to give compounds 3–5. Re2(CO)10 reacts differently. The different reduction potential of Re2(CO)10 and Mn2(CO)10 may be one reason.28 By employing [(DippForm)2LnII(thf)2] an unexpected [Re2(CO)8]2− anion is formed. In the case of [(Cp*)2SmII(thf)2], an unusual tetramerization of CO combined with the formation of a cyclic double Fischer-carbene type complex was seen. From previous work, we know that the ligand on the divalent samarium atom strongly influences the reactivity of the samarium reagent due to steric and electronic effects.
RY and PWR acknowledge funding for the current project from the SFB 1176 funded by the German Research Council (DFG). TS thanks the Alexander von Humboldt Foundation for a postdoctoral fellowship. Prof. Esther Roesch and Mr. Benjamin Basler are acknowledged for designing the cover artwork.
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Kölbel and M. Ralek, Catal. Rev., 1980, 21, 225 CrossRef.
- H. Mahmoudi, M. Mahmoudi, O. Doustdar, H. Jahangiri, A. Tsolakis, S. Gu and M. L. Wyszynski, Biofuels Eng., 2017, 2, 11 Search PubMed.
- R. Guettel, U. Kunz and T. Turek, Chem. Eng. Technol., 2008, 31, 746 CrossRef CAS.
- (a) B. B. Wayland, A. E. Sherry and V. L. Coffin, J. Chem. Soc., Chem. Commun., 1989, 662 RSC; (b) H. R. Sharpe, A. M. Geer, L. J. Taylor, B. M. Gridley, T. J. Blundell, A. J. Blake, E. S. Davies, W. Lewis, J. McMaster, D. Robinson and D. L. Kays, Nat. Commun., 2018, 9, 3757 CrossRef PubMed; (c) R. Y. Kong and M. R. Crimmin, J. Am. Chem. Soc., 2018, 140, 13614 CrossRef CAS PubMed; (d) P. J. Fagan, K. G. Moloy and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 6959 CrossRef CAS; (e) C. C. Cummins, G. D. Van Duyne, C. P. Schaller and P. T. Wolczanski, Organometallics, 1991, 10, 164 CrossRef CAS; (f) E. L. Werkema, L. Maron, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2007, 129, 2529 CrossRef CAS PubMed; (g) M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahon and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036 CrossRef CAS PubMed; (h) R. Lalrempuia, C. E. Kefalidis, S. J. Bonyhady, B. Schwarze, L. Maron, A. Stasch and C. Jones, J. Am. Chem. Soc., 2015, 137, 8944 CrossRef CAS PubMed.
- (a) O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, Science, 2006, 311, 829 CrossRef CAS PubMed; (b) N. Tsoureas, O. T. Summerscales, F. G. N. Cloke and S. M. Roe, Organometallics, 2013, 32, 1353 CrossRef CAS.
- O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, J. Am. Chem. Soc., 2006, 128, 9602 CrossRef CAS PubMed.
- K. G. Moloy, P. J. Fagan, J. M. Manriquez and T. J. Marks, J. Am. Chem. Soc., 1986, 108, 56 CrossRef CAS.
- W. J. Evans, D. S. Lee, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2006, 128, 14176 CrossRef CAS PubMed.
- W. J. Evans, A. L. Wayda, W. E. Hunter and J. L. Atwood, J. Chem. Soc., Chem. Commun., 1981, 706 RSC.
- B. Wang, G. Luo, M. Nishiura, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2017, 139, 16967 CrossRef CAS PubMed.
- T. Watanabe, Y. Ishida, T. Matsuo and H. Kawaguchi, J. Am. Chem. Soc., 2009, 131, 3474 CrossRef CAS PubMed.
- (a) M. J. Bennett, W. A. G. Graham, R. A. Smith and R. P. Stewart, J. Am. Chem. Soc., 1973, 95, 1684 CrossRef CAS; (b) B. Blank, M. Colling-Hendelkens, C. Kollann, K. Radacki, D. Rais, K. Uttinger, G. R. Whittell and H. Braunschweig, Chem. – Eur. J., 2007, 13, 4770 CrossRef CAS PubMed.
- G. B. Deacon, Z. Guo, P. C. Junk and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 8486 CrossRef CAS PubMed.
- (a) E. E. Benson and C. P. Kubiak, Chem. Commun., 2012, 48, 7374 RSC; (b) J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283 CrossRef CAS PubMed; (c) S. Oh, J. R. Gallagher, J. T. Miller and Y. Surendranath, J. Am. Chem. Soc., 2016, 138, 1820 CrossRef CAS PubMed.
- W. J. Evans, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1981, 103, 6507 CrossRef CAS.
- C. E. Kefalidis, S. Essafi, L. Perrin and L. Maron, Inorg. Chem., 2014, 53, 3427 CrossRef CAS PubMed.
- M. R. Churchill, K. N. Amoh and H. J. Wasserman, Inorg. Chem., 1981, 20, 1609 CrossRef CAS.
- L. L. Padolik, J. C. Gallucci and A. Wojcicki, J. Am. Chem. Soc., 1993, 115, 9986 CrossRef CAS.
- (a) C. E. Plečnik, S. Liu and S. G. Shore, Acc. Chem. Res., 2003, 36, 499 CrossRef PubMed; (b) T. D. Tilley and R. A. Andersen, J. Chem. Soc., Chem. Commun., 1981, 985 RSC; (c) P. V. Poplaukhin, X. Chen, E. A. Meyers and S. G. Shore, Inorg. Chem., 2006, 45, 10115 CrossRef CAS PubMed; (d) J. M. Boncella and R. A. Andersen, Inorg. Chem., 1984, 23, 432 CrossRef CAS; (e) T. D. Tilley and R. Andersen, J. Am. Chem. Soc., 1982, 104, 1772 CrossRef CAS; (f) C. E. Plečnik, S. Liu, X. Chen, E. A. Meyers and S. G. Shore, J. Am. Chem. Soc., 2004, 126, 204 CrossRef PubMed; (g) A. C. Hillier, A. Sella and M. R. J. Elsegood, J. Organomet. Chem., 1999, 588, 200 CrossRef CAS.
- (a) N. Arleth, M. T. Gamer, R. Köppe, S. N. Konchenko, M. Fleischmann, M. Scheer and P. W. Roesky, Angew. Chem., Int. Ed., 2016, 55, 1557 CrossRef CAS PubMed; (b) Y.-Z. Ma, S. Bestgen, M. T. Gamer, S. N. Konchenko and P. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 13249 CrossRef CAS PubMed; (c) C. Schoo, S. Bestgen, R. Köppe, S. N. Konchenko and P. W. Roesky, Chem. Commun., 2018, 54, 4770 RSC; (d) C. Schoo, S. Bestgen, M. Schmidt, S. N. Konchenko, M. Scheer and P. W. Roesky, Chem. Commun., 2016, 52, 13217 RSC.
- M. L. Cole and P. C. Junk, Chem. Commun., 2005, 2695 CAS.
- J. A. Baus, J. Poater, F. M. Bickelhaupt and R. Tacke, Eur. J. Inorg. Chem., 2017, 186 CrossRef CAS.
- (a) S.-M. Kuang, P. E. Fanwick and R. A. Walton, Inorg. Chem., 2001, 40, 5682 CrossRef CAS PubMed; (b) C. P. Casey, H. Sakaba, P. N. Hazin and D. R. Powell, J. Am. Chem. Soc., 1991, 113, 8165 CrossRef CAS; (c) B. Xu, Q.-s. Li, Y. Xie, R. B. King and H. F. Schaefer, Inorg. Chem., 2008, 47, 6779 CrossRef CAS PubMed.
- R. Heinzmann and R. Ahlrichs, Theor. Chim. Acta, 1976, 42, 33 CrossRef CAS.
- R. Ahlrichs and C. Ehrhardt, Chem. Unserer Zeit, 1985, 19, 120 CrossRef CAS.
- (a) R. F. W. Bader, Atoms in molecules: a quantum theory, Clarendon Press, Oxford, 1990 Search PubMed; (b) C. F. Matta and R. J. Gillespie, J. Chem. Educ., 2002, 79, 1141 CrossRef CAS.
- M. L. Cole, G. B. Deacon, C. M. Forsyth, P. C. Junk, K. Konstas, J. Wang, H. Bittig and D. Werner, Chem. – Eur. J., 2013, 19, 1410 CrossRef CAS PubMed.
- R. E. Dessy, P. M. Weissman and R. L. Pohl, J. Am. Chem. Soc., 1966, 88, 5117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterisation details, IR spectra and XRD data. CCDC 1905031–1905035. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc02350j |
| This journal is © The Royal Society of Chemistry 2019 |