
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the limits of Q-tag bioconjugation of antibodies†
Cristina
Marculescu
 ab,
Abirami
Lakshminarayanan‡
ab,
Joseph
Gault‡
b,
James C.
Knight
a,
Lisa K.
Folkes
a,
Thomas
Spink
a,
Carol V.
Robinson
b,
Katherine
Vallis
*a,
Benjamin G.
Davis
*b and
Bart
Cornelissen
*a
ab,
Abirami
Lakshminarayanan‡
ab,
Joseph
Gault‡
b,
James C.
Knight
a,
Lisa K.
Folkes
a,
Thomas
Spink
a,
Carol V.
Robinson
b,
Katherine
Vallis
*a,
Benjamin G.
Davis
*b and
Bart
Cornelissen
*a
aCRUK/MRC Oxford Institute for Radiation Oncology, Department of Oncology, University of Oxford, Oxford, OX3 7DQ, UK. E-mail: kathrine.vallis@oncology.ox.ac.uk; bart.cornelissen@oncology.ox.ac.uk
bChemistry Research Laboratory, University of Oxford, Oxford, OX1 3TA, UK. E-mail: ben.davis@chem.ox.ac.uk
First published on 3rd September 2019
Abstract
Site-selective labelling of antibodies (Abs) can circumvent problems from heterogeneity of conventional conjugation. Here, we evaluate the industrially-applied chemoenzymatic ‘Q-tag’ strategy based on transglutaminase-mediated (TGase) amide-bond formation in the generation of 89Zr-radiolabelled antibody conjugates. We show that, despite previously suggested high regioselectivity of TGases, in the anti-Her2 Ab Herceptin™ more precise native MS indicates only 70–80% functionalization at the target site (Q298H), in competition with modification at other sites, such as Q3H critically close to the CDR1 region.
Labelled antibodies (Abs) are vital clinical imaging tools and therapeutic agents.1 Generating conjugated Abs through site-specific conjugations that are more homogenously modified to clinically relevant standards is essential for future therapeutic use.2–4 Chemoenzymatic approaches can exploit the chemoselectivity and possible regioselectivity of even native residues in antibodies and can therefore enable ‘remodelling’ of existing antibodies.5–8 Transglutaminase (TGase) is one such enzyme that has been suggested to catalyze transamidation reactions of glutamine (Q) residues in a recognition sequence (the ‘Q-tag’) over other glutamines in heavy chains of IgGs, thus facilitating possible site-specific modification.9–11 As a consequence, TGase-mediated ‘Q-tag’ modification of Abs has been widely explored to generate Ab–drug conjugates,12,13 as well as labelled Abs,9,10,12,14 in both academia and industry.
Radiolabelled Abs find use in diagnostic imaging via e.g. Positron Emission Tomography (PET) or Single Photon Emission Computed Tomography (SPECT) as well as enabling great progress in immunotherapy.15 Zirconium-89 in particular has emerged as a powerful isotope for such applications. Its favorable half-life (∼3.3 days) is compatible with the slow clearance rate of Abs in vivo, allowing longer imaging whilst also providing high PET resolution.1689Zr-labelled Abs therefore represent a key demonstration system. Typically, 89Zr-labelled Abs are generated by initial conjugation of a suitable metal-chelator (e.g. siderophore deferoxamine (DFO)17) followed by radio-metal chelation.18 With few exceptions19–21 attachment of a metal ion chelator to Ab has been achieved by targeting nucleophilic ε-amines of several lysine (Lys) residues (Fig. 1A),22 resulting in heterogeneity.
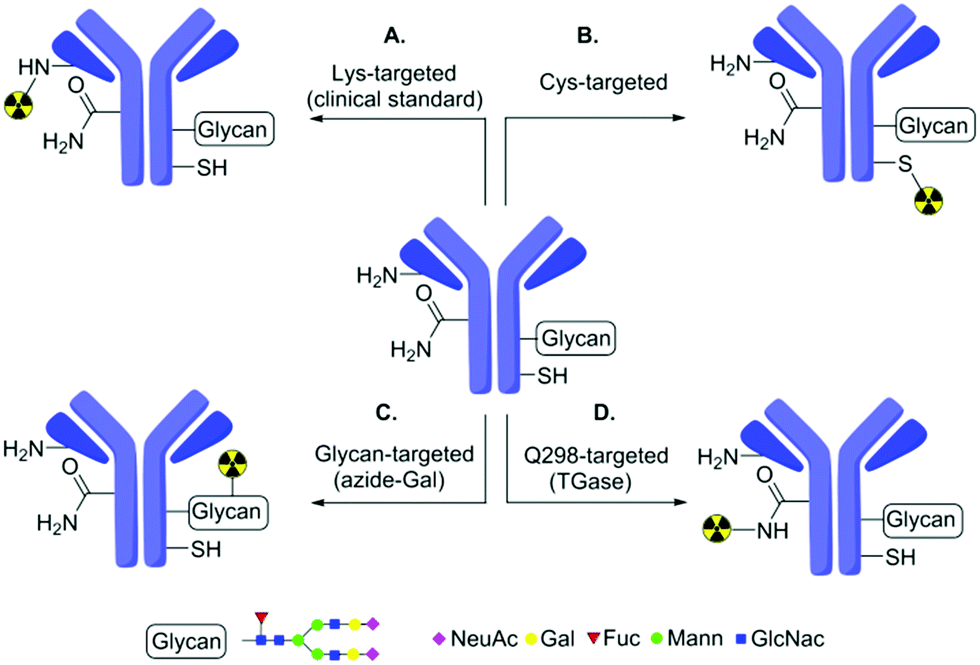 | ||
| Fig. 1 Strategies for 89Zr-radiolabelling of Abs. Traditional modification methods based on Lys (A) typically generate heterogeneity. To reduce heterogeneity, these have been extended by methods based on Cys (B), glycans (C) or glutamine targeting (D) using chemical or chemoenzymatic methods. (D) The ‘Q-tag’ system explored in this work has been previously proposed to be exclusively selective for Q298H in antibodies. | ||
To improve homogeneity, protein engineering can be combined with chemical modification to install and more-selectively label additional cysteine (Cys) residues (Fig. 1B).19 Alternatively, chemoenzymatic approaches can also be used to modify glycan residues on Ab (Fig. 1C). Whilst these can reduce heterogeneity compared to traditional methods, they may still yield partial heterogeneity due to, e.g. mixed glycosylation patterns or incomplete loading. Here, we show that an alternative, industrially-applied, chemoenzymatic method – the ‘Q-tag’ system (Fig. 1D)23 – allows successful generation of 89Zr-labelled Abs. Notably, whilst this improves homogeneity, our study also reveals previously unappreciated limits of Q-tag site-selectivity at sites likely to directly impair function.
The transamidation activity of TGases, which naturally cross-link Gln and Lys side-chains23 has been exploited previously to modify several proteins24 including in generation of Ab–drug (and other) conjugates.25–27 This method relies on a presumed high, but in fact rarely fully-characterized selectivity for certain peptide sequences containing Gln (so-called ‘Q-tags’). Amongst these is the sequence PWEEQYNST11 in IgG Abs containing a target Q298H residue (Herceptin numbering), found close to the N-glycosylated N300H (Fig. 1D). As a consequence of the glycosylation at site N300, site Q298 is typically sterically-occluded but can be revealed by prior treatment with the amidase PNGase, which converts glycosylated-N300 to D300 (Fig. 2).28
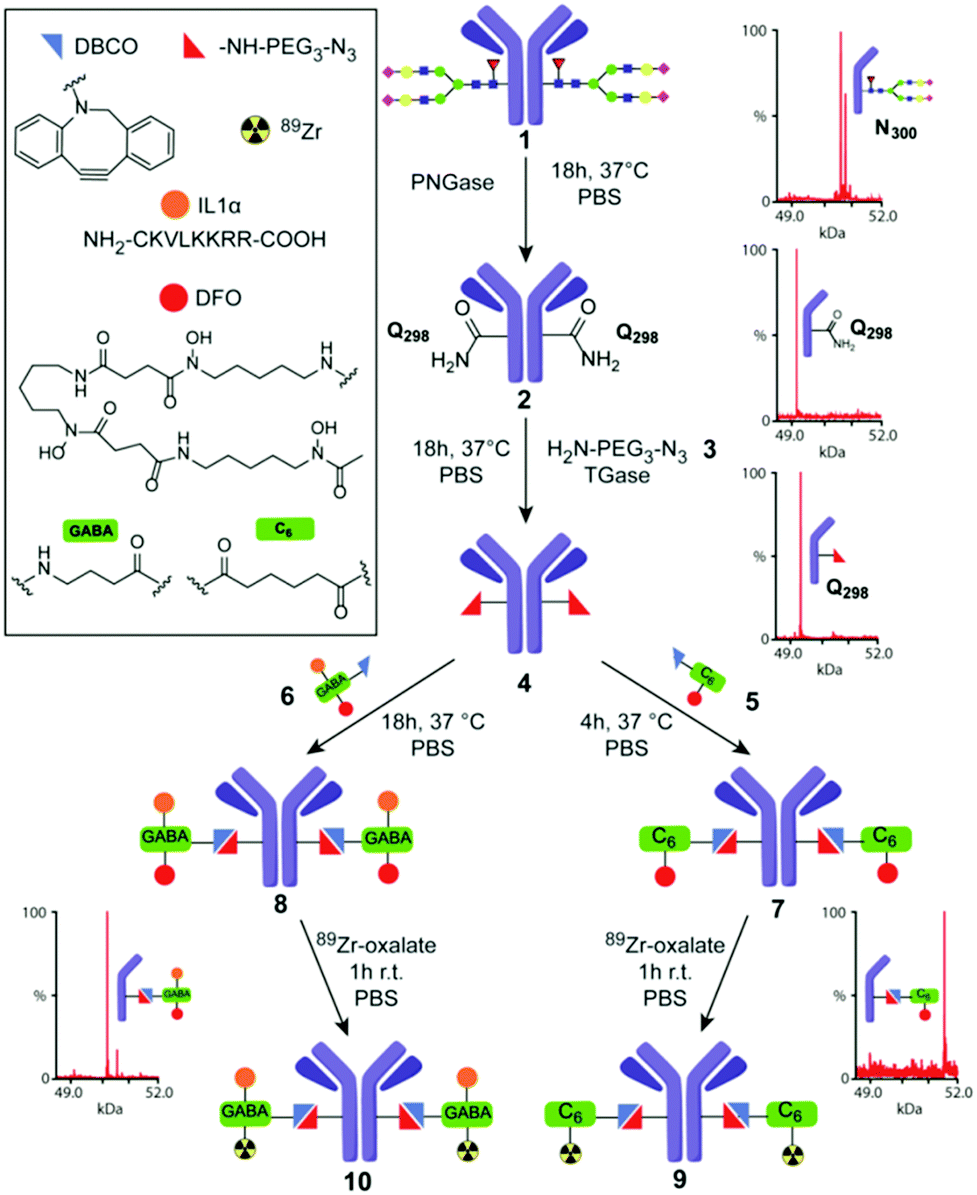 | ||
| Fig. 2 TGase-mediated, chemoenzymatic generation of 89Zr–Herceptin conjugates. A modular strategy based on ‘Q-tag’ allowed incorporation of variable moieties (see box). Sequential, chemoenzymatic remodeling using PNGase and TGase and then chemical conjugation was directly monitored by corresponding MS of heavy chain obtained under reducing, denaturing conditions (rLCMS) prior to final 89Zr chelation (see also ESI,† Scheme S1). | ||
We applied a combined PNGase/TGase modification method to generate 89Zr-labelled Abs with reduced heterogeneity in the classical anti-Her2 Herceptin™ system. We designed a modular process that would allow near-direct comparison with prior results,29 through attachment of a DFO chelator to allow radiolabelling with 89Zr. This also enabled additional modification with other functional moieties (Fig. 2). To avoid metal-mediated conjugation strategies, which might inhibit/interfere with DFO chelation, we chose strain-promoted triazole formation30 for conjugation with a PEG–azidoamine10 as primary-amine co-substrate for TGase (Fig. 2).
The protein substrate, deglycosylated (dg) Herceptin (dg-Her, 2), was generated by treating wild-type (wt) Herceptin (wt-Her, 1) with PNGaseF,31 creating 2 as a D300H Asp-variant of Her. Nearby and now accessible Q298H of dg-Her 2 was then conjugated to the azidoamine H2N–CH2CH2–(OCH2CH2)2–N3 (3) using the TGase from Streptomyces mobaraensis to install an azide residue into the side chain of Q298H (creating azido-Her 4) for subsequent reaction with strained alkynes. Initial LCMS under reducing, denaturing conditions (rLCMS) and reducing SDS-PAGE analysis (ESI,† Table S1, method A), suggested that deglycosylation and azide-incorporation steps proceeded to completion, converting wt-Her 1 into desired products dg-Her 2 and then azido-Her 4 (Fig. 2). Retained reactivity of the azide moiety in azido-dg-Her 4 was confirmed using a Cy3-dye-containing alkyne (ESI,† Fig S1). Notably, no modification of the light chain was observed using these analytical methods (Fig. 3B). Together these traditional modes of analysis proved consistent with highly site-selective alterations guided by the Q-tag sequence, as previously proposed.
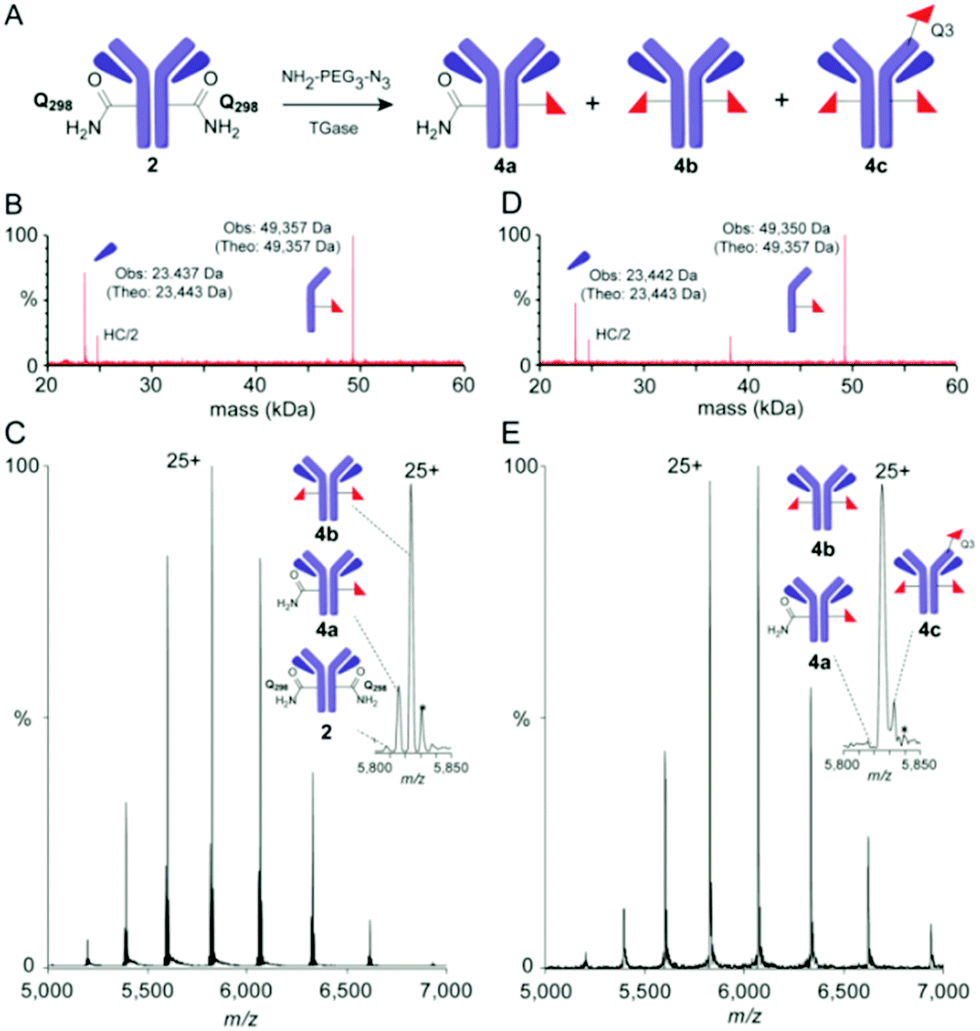 | ||
| Fig. 3 Precise monitoring of ‘Q-tag’ method reveals unexpected heterogeneity. (A) Reaction for TGase-mediated azide incorporation; (B) rLCMS and (C) nMS (spectrum and zoom into +25 charge state) of mixed azide-dg-Her 4 obtained using method A; (D) rLCMS and (E) nMS (spectrum and zoom of +25 charge state) of mixed azido-dg-Her 4 using method C reveals contaminant 4c bearing modification at Q3H. Note: nMS (C and E) also show additional species (*)33 assigned to sequence variations (+176 Da), consistent with prior analyses.6 | ||
Prior work by us and others6,32 has demonstrated that the heteromultimeric nature of monoclonal Abs can lead to misleading quantitative analyses via rLCMS and that high resolution native MS (nMS) of intact monoclonal antibody conjugates can provide more precision and accuracy. nMS of dg-Her 2 confirmed complete removal of N-glycans of wt-Her 1 (ESI,† Fig S2). However, analyses of azido-dg-Her 4 generated under various conditions (ESI,† Table S1)25 unexpectedly revealed mixtures of different Ab species with varied conjugation states (Fig. 3: 4a–c, azido copy numbers a = 1, b = 2, c = 3 plus unreacted 2). Together, these data revealed azido-dg-Her 4, formed under these conditions, is not homogeneous (Fig. 3C and E, also full MS data in ESI†).
Next, peptide mapping (tryptic-MS/MS) of azido-dg-Her 4 was used to dissect this surprising heterogeneity. This confirmed primary incorporation of modified residue azido-EG3-Q298H at target site Q298H (ESI,† Fig S3) could be achieved at levels of up to 70% (70.2% relative to 4b). Notably, however, it also revealed that attempts to drive further conversions (e.g. method C, higher concentrations of Ab, amine, TGase) instead gave triply-modified product 4c, bearing three azido moieties, at levels up to 14% (Fig. 3). Use of reduced equivalents of 3 gave only poor conversions (ESI,† Table S1, method E).
This was particularly surprising given the previously suggested selectivity10 of TGase for Q298H and for the ‘Q-tag’ sequence. Tryptic-MS/MS analysis of azido-Her 4 generated using method C allowed unambiguous identification of Q3H close to N-terminal CDR1 epitope-binding region as a third conjugation site (ESI,† Fig S4). Not only did this reveal limits of TGase-mediated ‘Q-tag’ conjugation, it also highlighted that the side-products contain modifications that may directly interfere with epitope binding due to proximity to CDRs.
Despite these unexpected and previously unappreciated limitations in the ‘Q-tag’, we were nonetheless able to generate useful target product mixtures (4) that were more homogeneous (∼80%) than those observed from typical chemical conjugations (e.g. via Lys – see ESI,† Fig. S7 for typical). This, in turn, allowed attachment of mono- and bi-functional moieties containing chelate DFO (using 5), or DFO + peptide (using 6). These modular DFO–alkyne 5 and DFO + IL1α-alkyne 6 reagents were themselves constructed using HATU-mediated amide bond formation (and maleimide conjugation in 6 – see ESI,† Scheme S1). Bi-functional 6 additionally contains a cell-penetrating peptide and nuclear localization sequence derived from hIL1α34 to test the introduction of a model peptide module that could allow interrogation of nuclear biomarkers in the future.35,36 These constructs and conjugations also allowed us to test the modularity of the TGase-based approach for building multi-functional Ab systems, by straightforward alteration of the corresponding alkyne-containing reaction partners. Reaction of 4 with both 5 or 6 proceeded with essentially full conversion (>95%), as judged by SDS-PAGE and rLCMS analysis (see ESI†), to yield conjugates [DFO]2-dg-Her 7 and [DFO + IL1α]2-dg-Her 8, respectively, with near-identical copy number distribution ∼2 (Fig. 2). This copy number distribution was also confirmed by nMS analysis with fully conjugated products as major species. Slight peak broadening due to adventitious DFO-metal binding reduced quantification precision by nMS, (ESI,† Fig. S5 and S6). Analysis of conjugate stability over prolonged periods suggested good stability for 7, but slow degradation of 8 (>6 months, via maleimide retro-Michael).
Radiolabelling of 7 and 8via chelation with 89Zr,37 yielded 89Zr-labelled dg-Her variants 89Zr·[DFO]2-dg-Her 9 and 89Zr·[DFO + IL1α]2-dg-Her 10 with radiochemical yields (RCY) of 94 ± 5% (n = 5) and 96 ± 5% (n = 7), respectively (Fig. 2). To allow side-by-side comparison with 89Zr–Herceptin conjugates obtained through conventional, random Lys-directed modification38,39 we also generated37 [DFO]mix-Her 11 (ESI,† Fig. S7). In contrast to site-selectively DFO-modified 8 and 9, rLCMS analysis of [DFO]mix-Her 11 indicated high heterogeneity in both heavy and light chains (ESI,† Fig. S7). Radiolabelling of 11 with 89Zr provided 89Zr·[DFO]mix-Her 12 in RCY up to 98%.37
Retained biological functions of these Herceptin™ conjugates 7, 8, 11 were evaluated through determination of in vitro binding affinities (KD) to Her2 using a saturation-binding assay (ESI,† Fig. S8) and were not significantly different (P < 0.05) from wt Herceptin™. Importantly, 89Zr·[DFO + IL1α]2-dg-Her 10 proved highly stable in human serum at 37 °C, retaining radiolabel even after 4 days of incubation (ESI,† Fig. S9) and suggesting promising suitability for future in vivo use.
We have shown ‘Q-tag’ TGase-mediated Ab-conjugation yields less homogeneous conjugates than previously thought. This, the first precise analyses of intact TGase-generated Ab-conjugates conducted with nMS, reveals limitations in selectivity of widely-applied TGase. In the case of Herceptin, side products were formed with unwanted modification at sites critically close to CDRs. In preliminary experiments with murine anti-γH2AX antibody, (ESI,† Fig. S10), these limits of regioselectivity in the ‘Q-tag’-TGase method appear to be similar or worse. Our results were obtained with single amine 3 deemed efficient in prior studies;11 other amines may display altered selectivity. Indeed, unexpected TGase-driven modification of human proteins with endogenous amines has recently been noted,40 further highlighting the implications of TGase plasticity with respect to amine and protein substrates.
Not withstanding these limitations, the method does allow the creation of variants with improved homogeneity (∼80%) over traditional bioconjugations and enables a modular approach, described here, with potential for adding multiple functionalities in chelating moiety without any apparent gross effect on function. Surprisingly, prior in vivo comparisons20 have suggested that there are no differences between random attachment methods and more selective methods; future work will probe in vivo benefits of reduced heterogeneity.
The authors acknowledge support from CRUK (C5255/A15935), MRC (MC_PC_12004), and CRUK/EPSRC Cancer Imaging Centre Oxford (C5255/A16466). C. V. R. is supported by Wellcome Trust Investigator Award (104633/Z/14/Z), ERC Advanced Grant (641317) and MRC programme grant (MR/N020413/1). A. L. acknowledges funding by CRUK (CRUKDF 0318-AL). J. G. is a Junior Research Fellow at The Queen's College. We thank M. Mosley for assistance.
Conflicts of interest
There are no conflicts of interest to declare.Notes and references
- A. C. Freise and A. M. Wu, Mol. Immunol., 2015, 67, 142–152 CrossRef CAS PubMed.
- P. Strop, S.-H. Liu, M. Dorywalska, K. Delaria, Russell G. Dushin, T.-T. Tran, W.-H. Ho, S. Farias, Meritxell G. Casas, Y. Abdiche, D. Zhou, R. Chandrasekaran, C. Samain, C. Loo, A. Rossi, M. Rickert, S. Krimm, T. Wong, Sherman M. Chin, J. Yu, J. Dilley, J. Chaparro-Riggers, Gary F. Filzen, C. J. O’Donnell, F. Wang, Jeremy S. Myers, J. Pons, David L. Shelton and A. Rajpal, Chem. Biol., 2013, 20, 161–167 CrossRef CAS PubMed.
- T. Liu, J. Du, X. Luo, P. G. Schultz and F. Wang, Curr. Opin. Chem. Biol., 2015, 28, 66–74 CrossRef CAS PubMed.
- B.-Q. Shen, K. Xu, L. Liu, H. Raab, S. Bhakta, M. Kenrick, K. L. Parsons-Reponte, J. Tien, S.-F. Yu, E. Mai, D. Li, J. Tibbitts, J. Baudys, O. M. Saad, S. J. Scales, P. J. McDonald, P. E. Hass, C. Eigenbrot, T. Nguyen, W. A. Solis, R. N. Fuji, K. M. Flagella, D. Patel, S. D. Spencer, L. A. Khawli, A. Ebens, W. L. Wong, R. Vandlen, S. Kaur, M. X. Sliwkowski, R. H. Scheller, P. Polakis and J. R. Junutula, Nat. Biotechnol., 2012, 30, 184 CrossRef CAS PubMed.
- R. Jefferis, Nat. Rev. Drug Discovery, 2009, 8, 226–234 CrossRef CAS PubMed.
- T. B. Parsons, W. B. Struwe, J. Gault, K. Yamamoto, T. A. Taylor, R. Raj, K. Wals, S. Mohammed, C. V. Robinson, J. L. P. Benesch and B. G. Davis, Angew. Chem., Int. Ed., 2016, 55, 2361–2367 CrossRef CAS PubMed.
- B. E. Cook, P. Adumeau, R. Membreno, K. E. Carnazza, C. Brand, T. Reiner, B. J. Agnew, J. S. Lewis and B. M. Zeglis, Bioconjugate Chem., 2016, 27, 1789–1795 CrossRef CAS PubMed.
- R. van Geel, M. A. Wijdeven, R. Heesbeen, J. M. M. Verkade, A. A. Wasiel, S. S. van Berkel and F. L. van Delft, Bioconjugate Chem., 2015, 26, 2233–2242 CrossRef CAS PubMed.
- S. Jeger, K. Zimmermann, A. Blanc, J. Grünberg, M. Honer, P. Hunziker, H. Struthers and R. Schibli, Angew. Chem., Int. Ed., 2010, 49, 9995–9997 CrossRef CAS PubMed.
- P. Dennler, A. Chiotellis, E. Fischer, D. Brégeon, C. Belmant, L. Gauthier, F. Lhospice, F. Romagne and R. Schibli, Bioconjugate Chem., 2014, 25, 569–578 CrossRef CAS PubMed.
- P. Strop, Bioconjugate Chem., 2014, 25, 855–862 CrossRef CAS PubMed.
- T. L. Mindt, V. Jungi, S. Wyss, A. Friedli, G. Pla, I. Novak-Hofer, J. Grünberg and R. Schibli, Bioconjugate Chem., 2008, 19, 271–278 CrossRef CAS PubMed.
- F. Lhospice, et al. , Mol. Pharmaceutics, 2015, 12, 1863–1871 CrossRef CAS PubMed.
- P. R. Spycher, C. A. Amann, J. E. Wehrmuller, D. R. Hurwitz, O. Kreis, D. Messmer, A. Ritler, A. Kuchler, A. Blanc, M. Behe, P. Walde and R. Schibli, ChemBioChem, 2017, 18, 1923–1927 CrossRef CAS PubMed.
- I. Colombo, M. Overchuk, J. Chen, R. M. Reilly, G. Zheng and S. Lheureux, Methods, 2017, 130, 23–35 CrossRef CAS PubMed.
- M. A. Deri, B. M. Zeglis, L. C. Francesconi and J. S. Lewis, Nucl. Med. Biol., 2013, 40, 3–14 CrossRef CAS PubMed.
- P. Adumeau, S. K. Sharma, C. Brent and B. M. Zeglis, Mol. Imaging Biol., 2016, 18, 153–165 CrossRef CAS PubMed.
- F. C. J. van de Watering, M. Rijpkema, L. Perk, U. Brinkmann, W. J. G. Oyen and O. C. Boerman, BioMed Res. Int., 2014, 2014, 203601 Search PubMed.
- J. L. Houghton, B. M. Zeglis, D. Abdel-Atti, R. Aggeler, R. Sawada, B. J. Agnew, W. W. Scholz and J. S. Lewis, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15850–15855 CrossRef CAS PubMed.
- J. N. Tinianow, H. S. Gill, A. Ogasawara, J. E. Flores, A. N. Vanderbilt, E. Luis, R. Vandlen, M. Darwish, J. R. Junutula, S.-P. Williams and J. Marik, Nucl. Med. Biol., 2010, 37, 289–297 CrossRef CAS PubMed.
- B. M. Zeglis, C. B. Davis, D. Abdel-Atti, S. D. Carlin, A. Chen, R. Aggeler, B. J. Agnew and J. S. Lewis, Bioconjugate Chem., 2014, 25, 2123–2128 CrossRef CAS PubMed.
- J. M. Lambert and R. V. J. Chari, J. Med. Chem., 2014, 57, 6949–6964 CrossRef CAS PubMed.
- L. Lorand and R. M. Graham, Nat. Rev. Mol. Cell Biol., 2003, 4, 140–156 CrossRef CAS PubMed.
- A. Nilo, I. Passalacqua, M. Fabbrini, M. Allan, A. Usera, F. Carboni, B. Brogioni, A. Pezzicoli, J. Cobb, M. R. Romano, I. Margarit, Q.-Y. Hu, F. Berti and R. Adamo, Bioconjugate Chem., 2015, 26, 1839–1849 CrossRef CAS PubMed.
- P. Dennler, L. K. Bailey, P. R. Spycher, R. Schibli and E. Fischer, ChemBioChem, 2015, 16, 861–867 CrossRef CAS PubMed.
- S. Jeger, K. Zimmermann, A. Blanc, J. Grünberg, M. Honer, P. Hunziker, H. Struthers and R. Schibli, Angew. Chem., Int. Ed., 2010, 49, 9995–9997 CrossRef CAS PubMed.
- S. Puthenveetil, S. Musto, F. Loganzo, L. N. Tumey, C. J. O’Donnell and E. Graziani, Bioconjugate Chem., 2016, 27, 1030–1039 CrossRef CAS PubMed.
- J. Khoshnoodi, S. Hill, K. Tryggvason, B. Hudson and D. B. Friedman, J. Mass Spectrom., 2007, 42, 370–379 CrossRef CAS PubMed.
- X. Li, T. Fang and G. J. Boons, Angew. Chem., Int. Ed., 2014, 53, 7179–7182 CrossRef CAS PubMed.
- M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC.
- G. E. Norris, A. J. Flaus, C. H. Moore and E. N. Baker, J. Mol. Biol., 1994, 241, 624–626 CrossRef CAS PubMed.
- A. Beck, F. Debaene, H. Diemer, E. Wagner-Rousset, O. Colas, A. V. Dorsselaer and S. Cianférani, J. Mass Spectrom., 2015, 50, 285–297 CrossRef CAS PubMed.
- D. Nebija, H. Kopelent-Frank, E. Urban, C. R. Noe and B. Lachmann, J. Pharm. Biomed. Anal., 2011, 56, 684–691 CrossRef CAS PubMed.
- J.-H. Koo, H. Yoon, W.-J. Kim, S. Lim, H.-J. Park and J.-M. Choi, Mol. Biol. Rep., 2014, 41, 8117–8126 CrossRef CAS PubMed.
- B. Cornelissen, V. Kersemans, S. Darbar, J. Thompson, K. Shah, K. Sleeth, M. A. Hill and K. A. Vallis, Cancer Res., 2011, 71, 4539–4549 CrossRef CAS PubMed.
- M. Jain, S. C. Chauhan, A. P. Singh, G. Venkatraman, D. Colcher and S. K. Batra, Cancer Res., 2005, 65, 7840–7846 CrossRef CAS PubMed.
- J. C. Knight, S. J. Paisey, A. M. Dabkowski, C. Marculescu, A. S. Williams, C. Marshall and B. Cornelissen, Dalton Trans., 2016, 45, 6343–6347 RSC.
- E. C. F. Dijkers, J. G. W. Kosterink, A. P. Rademaker, L. R. Perk, G. A. M. S. van Dongen, J. Bart, J. R. de Jong, E. G. E. de Vries and M. N. Lub-deHooge, J. Nucl. Med., 2009, 50, 974–981 CrossRef CAS PubMed.
- S. B. M. Gaykema, C. P. Schröder, J. Vitfell-Rasmussen, S. Chua, T. H. Oude Munnink, A. H. Brouwers, A. H. H. Bongaerts, M. Akimov, C. Fernandez-Ibarra, M. N. Lub-deHooge, E. G. E. de Vries, C. Swanton and U. Banerji, Clin. Cancer Res., 2014, 20, 3945–3954 CrossRef CAS PubMed.
- L. A. Farrelly, et al. , Nature, 2019, 567, 535–539 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods section including the synthesis of Ab-labelling reagents, Ab conjugation methods, chromatography and native MS. See DOI: 10.1039/c9cc02303h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |