
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Evidence for tetranuclear bis-μ-oxo cubane species in molecular iridium-based water oxidation catalysts from XAS analysis†
Stuart A.
Bartlett
 *a,
Emma V.
Sackville
b,
Emma K.
Gibson
ac,
Veronica
Celorrio
a,
Peter P.
Wells
ad,
Maarten
Nachtegaal
e,
Stafford W.
Sheehan
f and
Ulrich
Hintermair
*b
*a,
Emma V.
Sackville
b,
Emma K.
Gibson
ac,
Veronica
Celorrio
a,
Peter P.
Wells
ad,
Maarten
Nachtegaal
e,
Stafford W.
Sheehan
f and
Ulrich
Hintermair
*b
aDiamond Light Source, Harwell Science and Innovation Campus, Didcot OX11 0DE, UK. E-mail: stuart.bartlett@diamond.ac.uk
bCentre for Sustainable Chemical Technologies, University of Bath, Bath BA2 7AY, UK. E-mail: u.hintermair@bath.ac.uk
cSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK
dSchool of Chemistry, University of Southampton, Southampton SO17 1BJ, UK
ePaul Scherrer Institute, 5232 Villigen, Switzerland
fCatalytic Innovations, Fall River, MA, 02723, USA
First published on 11th June 2019
Abstract
The structure of a highly active pyridine-alkoxide iridium water oxidation catalyst (WOC) is examined by X-ray absorption spectroscopy (XAS). A detailed comparison with IrO2 points to a rigid molecular unit of low nuclearity, with the best analysis suggesting a novel tetrameric iridium-oxo cubane as the resting state.
The water oxidation reaction continues to present a major challenge for the efficient transformation of renewable electricity into chemical energy.1,2 Although a number of well-defined molecular precursors to highly active water oxidation catalysts (WOCs) have been developed,3,4 there still is a distinct lack of structural and electronic insight into their reaction intermediates. This often hampers the rational development toward improved systems that meet the robustness and stability requirements for practical applications. To date, commercial electrolysers thus exclusively employ heterogeneous metal oxide anodes, with amorphous iridium oxy-hydroxide still standing as the most active and durable WOC material known.5 A number of recent X-ray absorption spectroscopy (XAS), X-ray photoelectron spectroscopy (XPS), UV-vis spectroscopy, and electrochemistry studies of IrOx surfaces under working conditions consistently suggest IrIII–IrIV–IrV redox cycles across two neighboring metal sites to effect the four-electron oxidation of water to O2.6–11
The geometry of rutile IrO2 suggests this to be possible through a “diamond core” bis-μ-oxo unit, a structural feature that is also found in many other WOCs based on manganese,12 ruthenium13 and cobalt oxides,14 including the active site of the natural oxygen-evolving complex (OEC) in photosystem II.15 Over the past decade some highly active molecular Ir-based WOCs have been developed,16 though some have been shown to rapidly transform into IrOx nanoparticles in situ.17 Strongly binding and oxidatively robust ligands (such as pyridine-alkoxides18) however yield highly active WOC solutions that are fully homogeneous.19 After activation20 these can even be grafted onto conducting metal oxides as molecular monolayers to yield exceptionally efficient and durable anodes for electrochemical water oxidation.21–23 Although a number of spectroscopic features have been analyzed,24 the exact molecular structure of these highly active catalysts has not yet been elucidated. A previous study of a pyridine-alkoxide ligated Ir WOC using high-energy X-ray scattering (HEXS) analysis and density functional theory (DFT) modelling suggested a mono-μ-oxo Ir dimer as the resting state of the activated catalyst,25 and in the following a series of coordinately saturated mono-μ-oxo dimers with two pyalk ligands per iridium centre have been synthesised.26 These intriguing model compounds allowed the direct characterization of higher valent states not observable in the parent catalyst bearing one pyalk ligand per metal, but the question how relevant the mono-μ-oxo motif to catalysis is remains. Here we present our results from a detailed X-ray absorption spectroscopy study of a molecular Ir-pyalk WOC in solution that provide strong evidence for bis-μ-oxo units in dimeric and tetrameric species as the resting state of the catalyst.
XAS has been successfully employed in several cases to gain structural insight into functional WOCs, including the CaMn4 oxo-cubane OEC in PSII,27 CoOx films14,28 and molecular Ru WOCs immobilized on surfaces.29 Here we investigated the activated form of [Cp*Ir(dimethyl-pyridinealkoxide)Cl] obtained by stirring with 50 equivalents of NaIO4 in water at room temperature for 16 hours (termed Ir-pyalk WOC), conditions which have previously been shown to yield a homogeneous iridium species without any bound Cp* but retaining the pyalk ligand.19,24,30 XAS measurements were performed directly on freshly prepared 5 mM aqueous solutions, and on cellulose pellets of the dark blue solid obtained after slow evaporation of these solutions to dryness (yielding identical spectra). Reference samples of Ir0 (metal foil), IrI ([Ir(cod)Cl]2), IrIII ([Cp*Ir(pyalk)Cl] and [Cp*Ir(H2O)3]SO4), and IrIV (Na2[IrCl6] and IrO2) were measured as standards for the different metal oxidation states. The XAS data was collected at the SuperXAS beamline at the Swiss Light Source and B18 at the UK Diamond Light Source at the iridium L3 edge at 11–12.5 keV in fluorescence mode.
The comparative XANES of the reference samples (Fig. 1) showed a general trend of increasing white line intensity with increasing oxidation states. This is expected due to the Ir L3 edge X-ray absorption constituting an allowed 2p → 5d transition, where the intensity increases in higher oxidation states as more states become available in the 5d orbitals, but shielding dampens the effective nuclear charge so that transition energies are not systematically affected by the oxidation state.31,32 The Ir-pyalk WOC sample in question having very similar XANES to that of IrO2 suggested the catalyst to rest in the IrIV oxidation state, consistent with its characteristic blue color and previous XPS analysis.24
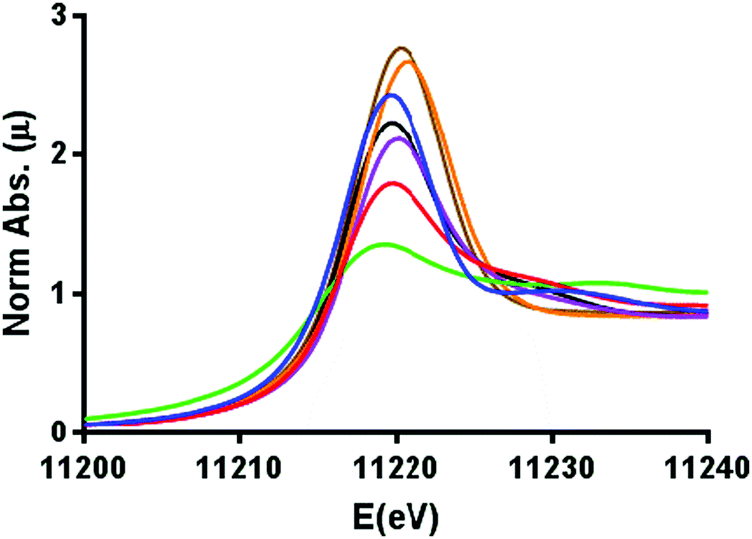 | ||
| Fig. 1 Normalized XANES spectra calibrated to the Pt0 L3 edge, displaying iridium L3 edge energies in Ir0 foil (green), [IrI(cod)Cl]2 (red), [Cp*IrIII(H2O)3]SO4 (purple), [Cp*IrIII(pyalk)Cl] (black), Na2[IrIVCl6] (blue), IrIVO2 (orange), and the Ir-pyalk WOC (brown). For further details see Fig. S5 (ESI†). | ||
Analysis of the extended X-ray absorption fine structure (EXAFS) however revealed distinct differences in their respective structures. Fig. 2 shows an overlay of the FT-EXAFS spectra of the IrO2 reference and the Ir-pyalk WOC sample, revealing a similar first coordination shell (blue shaded section) but major differences in the longer-range order between the two samples (green shaded section).
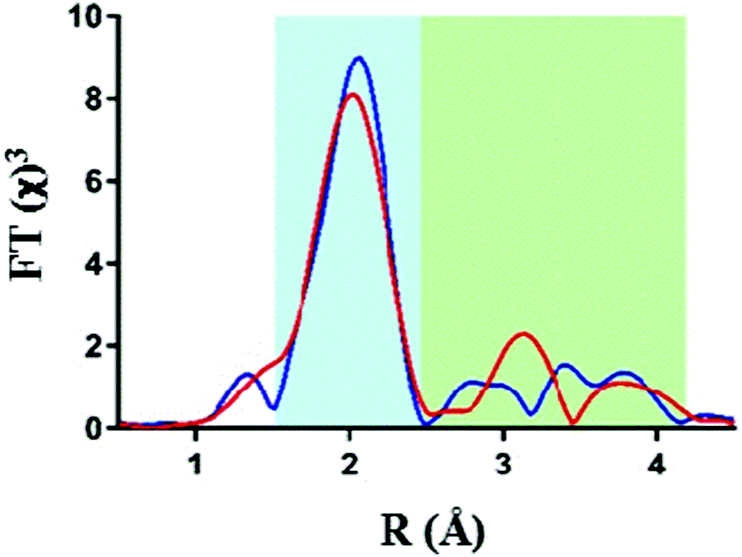 | ||
| Fig. 2 Comparison of Ir L3 edge EXAFS data with k3-weighting, showing Fourier transform EXAFS data for IrO2 (red) and the Ir-pyalk WOC (blue). | ||
The best fit parameters for the IrO2 EXAFS spectrum showed the first coordinating shell around 2 Å to consist of six bridging oxygen atoms with a uniform Ir–O bond distance of 1.98 Å (Table 1). The similarity of the first shell between IrO2 and Ir-pyalk WOC suggests a similar direct bonding environment to be present in the Ir-pyalk WOC sample (i.e., six low Z atoms around 2 Å). The analysis of peaks >2.5 Å of the IrO2 spectrum required defining each single contribution from the overlapping multiple backscattering pathways. These multiple scattering pathways are particularly prevalent in rigid, ordered structures such as crystalline metal oxides, but are known to be difficult to resolve for iridium due the absence of characteristic high amplitude peaks around 3 Å in the Fourier transform.33 A comprehensive fitting analysis yielded optimum parameters indicating two neighboring Ir atoms at 3.11 Å and eight Ir atoms at 3.52 Å (Table 1, Table S1 and Fig. S2, ESI†), fully consistent with XRD results.34
| Structure | Path | Distance (Å) | σ 2 (Å2) | R fac |
|---|---|---|---|---|
| IrO2 | 6 Ir–O | 1.98(1) | 0.006(1) | 0.001 |
| 2 Ir–Ir | 3.11(3) | 0.006(3) | ||
| 8 Ir–Ir | 3.52(8) | 0.017(11) | ||
| 1 | 6 Ir–O/N | 2.04 | 0.004 | 0.014 |
| 2 Ir–C | 2.81 | 0.009 | ||
| 4 Ir–C | 2.97 | 0.006 | ||
| 1 Ir–Ir | 3.56 | 0.002 | ||
| 2 | 2 Ir–O | 1.96(1) | 0.002 | 0.006 |
| 4 Ir–O/N | 2.50(7) | 0.012(13) | ||
| 1 Ir–Ir | 2.97(4) | 0.002(3) | ||
| 3 | 3 Ir–O | 1.96(3) | 0.006(4) | 0.001 |
| 3 Ir–O/N | 2.04(3) | 0.001(1) | ||
| 3 Ir–Ir | 2.97(3) | 0.012(3) |
To fit and compare the FT EXAFS spectrum of the Ir-pyalk WOC three structural motifs were investigated: a flexible mono-μ-oxo dimer (1),25 a more rigid bis-μ-oxo dimer (2),24 and a more extended “dimer of dimers” bis-μ-oxo cubane (3) (Fig. 3 and Table 1).
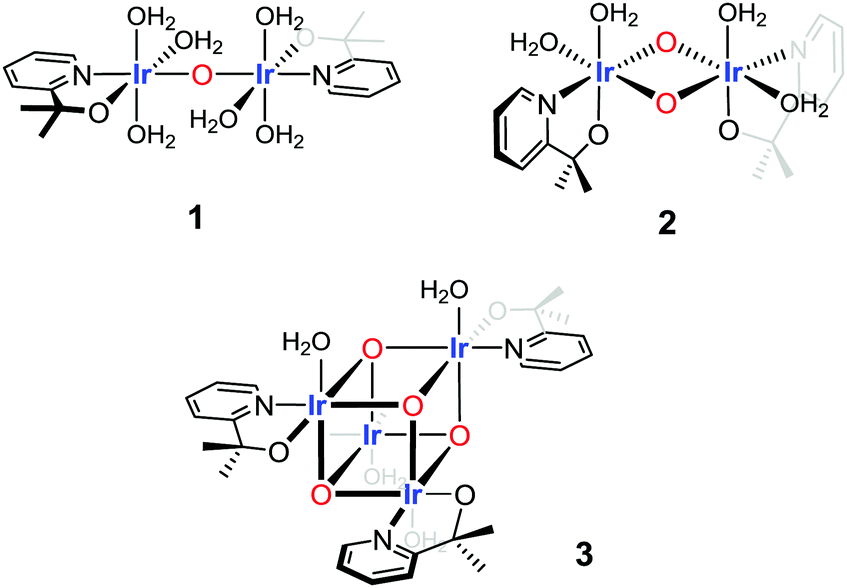 | ||
| Fig. 3 Three structural motifs used in the EXAFS fitting analysis of the Ir-pyalk WOC derived from [Cp*Ir(dimethyl-pyridinealkoxide)Cl] in dilute aqueous solution. | ||
The bis-μ-oxo dimer structure 2 gave a high statistical fit for a short Ir–Ir distance of 2.97 Å with two bridging oxygen atoms, distinctly shorter than the 3.11 Å found in the “diamond core” of IrO2 by XAS (Table 1) and XRD.34 This contraction mainly originated from coordination of the chelating pyalk ligand, and not from a switch to μ2-O from μ3-O in IrO2 (see also discussion of 3 below). With the four terminal N/O ligands from the pyalk ligand and coordinated water molecules forming the second shell, a good statistical fit with an Rfac (measure of fit accuracy, see ESI†) of 0.006 was obtained for 2 (Fig. 4 and Table S2, ESI†). All attempts to fit the flexible mono-μ-oxo dimer 125 to our EXAFS data yielded much poorer statistical fits of Rfac = 0.014 or higher. This arises due to the inclusion of a substantially increased Ir–Ir distance of 3.56 Å originating from the mono-μ-oxo unit. We found this to be incompatible with our EXAFS data showing that the Ir–Ir distance must be less than 3 Å, which cannot be obtained with a single oxo bridge between the metals.35
 | ||
| Fig. 4 Ir L3 edge EXAFS spectra of the Ir-pyalk WOC in solution (blue) comparing three simulated fit models from Fig. 3 (red dashed): mono-μ2-oxo dimer 1, bis-μ2-oxo dimer 2, and bis-μ3-oxo cubane 3. Displaying k3 weighted Fourier Transform (with imaginary part, left) and k3 weighted EXAFS plot (right) for each structure. | ||
On the other hand, the even more rigid cubane structure 3 with three Ir contributions maintained the same Ir–Ir distance as found in 2, with the remaining three N/O ligands moving to a shorter distance of 2.06(2) Å (Table S3 (ESI†) and Fig. 4). This arrangement provided an even better fit with a global Rfac of 0.001, showing excellent agreement with the EXAFS data out to k ranges beyond 10 Å−1 (Fig. 4).
Inspecting the individual pathway contributions in the best fit analysis between structure 3 and 2, only in the cubane structure 3 did the higher Ir coordination number adequately fit the shoulder peak contribution at 3.0 Å in the imaginary part of the Fourier Transform (Fig. S6, ESI†). In the dimer model 2 the Ir–Ir shell cannot fully reproduce this peak, requiring a shift to higher distance for the ligands to compensate, thus degrading the overall fit (Fig. S6, ESI†). This is indication that a nuclearity greater than two is present in the majority of the sample, with the best fitting analysis suggesting a neighboring iridium number of three as in the cubane structure 3. Indeed, setting the Ir–Ir coordination number as a floating variable, the fitting optimization converged to an average of 2.6(4) neighboring Ir atoms. In addition, literature XRD data36 are in good agreement with the IrIV–N/O distances of 2.05 Å found in 3.
Comparing structures 2 and 3, the cubane differs in the reduction of terminal ligand sites from four to three, a shortening of Ir–N/O distances from 2.51 Å to 2.04 Å, and an increase of neighboring Ir atoms from one to three in the tetranuclear oxo-cubane. This provided a superior statistical fit with realistic bond distances for structure 3 even compared to the good fit for structure 2. This analysis shows both bis-μ-oxo dimers and tetramers to be viable possibilities, with the EXAFS fit indicating an oxo-cubane type ‘dimer-of-dimer’ structure 3 to be the most realistic structural representation of the resting state of the activated Ir-pyalk WOC in aqueous solution.
We have provided a detailed structural analysis of a prominent molecular WOC through iridium L3 edge XAS. The results find mono-μ-oxo linkages to allow too much flexibility and induce too large Ir–Ir distances to be compatible with experimental EXAFS data of the resting state of the activated catalyst, suggesting that such species may not be relevant to water oxidation catalysis.35 Our analysis points to more compact and rigid bis-μ-oxo linkages in oxo-cubane ‘dimer-of-dimers’ as the key structural feature in these highly active and durable WOCs. Similar metal-oxo cubanes have been shown to feature in a number of WOCs based on other transition metals such as Mn, Co, and Ni (including the natural OEC in PSII),27,37 and their unique robustness and redox chemistry may represent a common design criterion allowing for high activity and stability in the demanding 4-electron oxidation of water. Importantly, a pyalk-modified bis-μ-oxo Ir4O4 cubane would be consistent with previously reported characterization data including 17O NMR, EPR, UV-vis and resonance-Raman spectroscopies,24 and shed new light on the mode of surface binding of these catalysts.38–40 However, we caution that the kinetic role of such tetrameric resting states will have to be clarified prior to mechanistic interpretations. Recently a half order in [Ir] on the rate of O2 evolution has been found for a range of pyalk-ligated Ir WOCs using NaIO4, implying a dominant dimeric resting state liberating small amounts of active monomer into the catalytic cycle.41 In light of the present findings this could mean a tetramer–dimer, or tetramer–dimer–monomer equilibria to be operational instead. Ascertaining how many metal sites are involved in the turnover-limiting step will be important for deciphering the workings of these highly active catalysts.
This work was supported by the Royal Society (UF160458; University Research Fellowship to UH), the EPSRC Centre for Doctoral Training in Sustainable Chemical Technologies (EP/L016354/1; PhD studentship to EVS), and the UK Catalysis Hub (EP/K014714/1). The authors acknowledge the Diamond Light Source for provision of beamtime (SP10306 and SP15151). The authors would like to thank Mark Styles and Nathan Webster (CSIRO, Australia), Robert Potter (Johnson Matthey), and Paul Frith (University of Bath) for their support and assistance with this project.
Conflicts of interest
US Patent No. 9,790,605 by SWS, UH et al. contains intellectual property described in this article. The other authors declare no competing financial interest.Notes and references
- A. Nicola and B. Vincenzo, Angew. Chem., Int. Ed., 2007, 46(1–2), 52 Search PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. Mayrhofer, Angew. Chem., Int. Ed., 2014, 53(1), 102 CrossRef CAS.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114(24), 11863 CrossRef.
- J. D. Blakemore, R. H. Crabtree and G. W. Brudvig, Chem. Rev., 2015, 115(23), 12974 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135(45), 16977 CrossRef CAS PubMed.
- P. Steegstra, M. Busch, I. Panas and E. Ahlberg, J. Phys. Chem. C, 2013, 117(40), 20975 CrossRef CAS.
- A. Minguzzi, C. Locatelli, O. Lugaresi, E. Achilli, G. Cappelletti, M. Scavini, M. Coduri, P. Masala, B. Sacchi, A. Vertova, P. Ghigna and S. Rondinini, ACS Catal., 2015, 5(9), 5104 CrossRef CAS.
- A. Minguzzi, O. Lugaresi, E. Achilli, C. Locatelli, A. Vertova, P. Ghigna and S. Rondinini, Chem. Sci., 2014, 5(9), 3591 RSC.
- H. G. S. Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, Angew. Chem., Int. Ed., 2014, 53(28), 7169 CrossRef PubMed.
- V. Pfeifer, T. E. Jones, S. Wrabetz, C. Massué, J. J. Velasco Vélez, R. Arrigo, M. Scherzer, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2016, 7(11), 6791 RSC.
- H. Ooka, Y. Wang, A. Yamaguchi, M. Hatakeyama, S. Nakamura, K. Hashimoto and R. Nakamura, Phys. Chem. Chem. Phys., 2016, 18(22), 15199 RSC.
- A. Bergmann, I. Zaharieva, H. Dau and P. Strasser, Energy Environ. Sci., 2013, 6(9), 2745 RSC.
- H. Over, Chem. Rev., 2012, 112(6), 3356 CrossRef CAS.
- M. Risch, V. Khare, I. Zaharieva, L. Gerencser, P. Chernev and H. Dau, J. Am. Chem. Soc., 2009, 131(20), 6936 CrossRef CAS PubMed.
- J. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106(11), 4455 CrossRef CAS PubMed.
- J. M. Thomsen, D. L. Huang, R. H. Crabtree and G. W. Brudvig, Dalton Trans., 2015, 44(28), 12452 RSC.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42(6), 2338 RSC.
- T. K. Michaelos, D. Y. Shopov, S. B. Sinha, L. S. Sharninghausen, K. J. Fisher, H. M. C. Lant, R. H. Crabtree and G. W. Brudvig, Acc. Chem. Res., 2017, 50(4), 952 CrossRef CAS PubMed.
- U. Hintermair, S. M. Hashmi, M. Elimelech and R. H. Crabtree, J. Am. Chem. Soc., 2012, 134(23), 9785 CrossRef CAS PubMed.
- J. M. Thomsen, S. W. Sheehan, S. M. Hashmi, J. Campos, U. Hintermair, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2014, 136(39), 13826 CrossRef CAS PubMed.
- S. W. Sheehan, J. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun., 2015, 6, 6469 CrossRef CAS PubMed.
- W. Li, S. W. Sheehan, D. He, Y. He, X. Yao, R. L. Grimm, G. W. Brudvig and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 11428 CrossRef CAS PubMed.
- J. W. Moir, E. V. Sackville, U. Hintermair and G. A. Ozin, J. Phys. Chem. C, 2016, 120(24), 12999 CrossRef CAS.
- U. Hintermair, S. W. Sheehan, A. R. Parent, D. H. Ess, D. T. Richens, P. H. Vaccaro, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2013, 135(29), 10837 CrossRef CAS PubMed.
- K. R. Yang, A. J. Matula, G. Kwon, J. Hong, S. W. Sheehan, J. M. Thomsen, G. W. Brudvig, R. H. Crabtree, D. M. Tiede, L. X. Chen and V. S. Batista, J. Am. Chem. Soc., 2016, 138(17), 5511 CrossRef CAS PubMed.
- L. S. Sharninghausen, S. B. Sinha, D. Y. Shopov, B. Choi, B. Q. Mercado, X. Roy, D. Balcells, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2016, 138(49), 15917 CrossRef CAS PubMed.
- J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni and V. K. Yachandra, Science, 2006, 314(5800), 821 CrossRef CAS PubMed.
- X. Li, E. B. Clatworthy, S. A. Bartlett, A. F. Masters and T. Maschmeyer, J. Phys. Chem. C, 2017, 121(21), 11021 CrossRef CAS.
- D. Lebedev, Y. Pineda-Galvan, Y. Tokimaru, A. Fedorov, N. Kaeffer, C. Copéret and Y. Pushkar, J. Am. Chem. Soc., 2018, 140(1), 451 CrossRef CAS PubMed.
- E. V. Sackville, G. Kociok-Köhn and U. Hintermair, Organometallics, 2017, 36(18), 3578 CrossRef CAS.
- A. Uzan and B. C. Gates, Angew. Chem., Int. Ed., 2008, 47(48), 9245 CrossRef PubMed.
- J. H. Choy, K. Dong-Kuk, G. Demazeau and D. Y. Jung, J. Phys. Chem., 1994, 98, 6258 CrossRef CAS.
- E. Prouzet, J. Phys.: Condens. Matter, 1995, 7, 8027 CrossRef CAS.
- A. A. Bolzan, C. Fong, B. J. Kennedy and C. J. Howard, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 373 CrossRef.
- A possible explanation for previous findings of Ir–Ir distances of ∼3.5 Å, leading to the proposal of mono-μ-oxo structures, could be the fact samples had been prepared at much higher Ir concentrations in advance of the analysis. This may have led to the formation of IrO2 particles that give Ir–Ir backscattering peaks in exactly that region (see Table 1), which we did not observe in homogeneous samples freshly prepared below the critical Ir concentration of 10 mM.
- S. B. Sinha, D. Y. Shopov, L. S. Sharninghausen, C. J. Stein, B. Q. Mercado, D. Balcells, T. Pedersen, M. Reiher, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2017, 139, 9672 CrossRef CAS PubMed.
- F. Song, R. Moré, M. Schilling, G. Smolentsev, N. Azzaroli, T. Fox, S. Luber and G. R. Patzke, J. Am. Chem. Soc., 2017, 139(40), 14198 CrossRef CAS PubMed.
- Y. Zhao, K. R. Yang, Z. Wang, X. Yan, S. Cao, Y. Ye, Q. Dong, X. Zhang, J. E. Thorne, L. Jin, K. L. Materna, A. Trimpalis, H. Bai, S. C. Fakra, X. Zhong, P. Wang, X. Pan, J. Guo, M. Flytzani-Stephanopoulos, G. W. Brudvig, V. S. Batista and D. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(12), 2902 CrossRef CAS PubMed.
- Y. Zhao, X. Yan, K. R. Yang, S. Cao, Q. Dong, J. E. Thorne, K. L. Materna, S. Zhu, X. Pan, M. Flytzani-Stephanopoulos, G. W. Brudvig, V. S. Batista and D. Wang, ACS Cent. Sci., 2018, 4(9), 1166 CrossRef CAS.
- I. Poli, U. Hintermair, M. Regue, S. Kumar, E. V. Sackville, J. Baker, T. M. Watson, S. Eslava and P. J. Cameron, Nat. Commun., 2019, 10, 2097 CrossRef PubMed.
- E. V. Sackville, F. Marken and U. Hintermair, ChemCatChem, 2018, 10(19), 4280 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of XAS fitting analyses (XANES and EXAFS), structural details and additional plots. See DOI: 10.1039/c9cc02088h |
| This journal is © The Royal Society of Chemistry 2019 |