
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-temperature synthesized Li4Mn5O12-like cathode with hybrid cation- and anion-redox capacities†
Yang
Liu
ab,
Guang
Liu
ab,
Hui
Xu
ab,
Yuheng
Zheng
ab,
Yunhui
Huang
ab,
Sa
Li
*ab and
Ju
Li
 *c
*c
aSchool of Materials Science and Engineering, Tongji University, Shanghai 201804, China. E-mail: lisa@tongji.edu.cn
bInstitute of New Energy for Vehicles, Tongji University, Shanghai 201804, China
cDepartment of Nuclear Science and Engineering and Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: liju@mit.edu
First published on 26th April 2019
Abstract
The Li-rich spinel Li4Mn5O12 (Li(Mn5/3Li1/3)O4) historically only shows a reversible cation-redox reaction, with a theoretical capacity of 135.5 mA h g−1. However, we found that a simple 400 °C solid-state synthesis method gives a Li4Mn5O12-like nanoparticulate cathode that yields significant reversible hybrid cation- and anion-redox capacities. A high specific capacity of 212 mA h g−1 was achieved. The reversible anion-redox contribution is attributed to the tiny particle size (<10 nm), which facilitates electron tunneling, and a possible random solid-solution in the Li(Mn5/3Li1/3)O4 lattice due to the low synthesis temperature.
Although the spinel Li4Mn5O12 is cost-effective1 and environment-friendly without the expensive and toxic Co or Ni, it has significant challenges to meet the demands of rechargeable batteries. Among them, the relatively low specific capacity has been a bottleneck ever since it was initially reported by M. M. Thackeray two decades ago.2,3 Starting with Li4+Mn54+O122−, because the Mn ions are already tetravalent, delithiation cannot occur4 without a concomitant change in the average oxygen valency as Li4−m+Mn54+O12(2−a)−, where a = m/12 ≥ 0 tracks the degree of oxygen anion-redox capacity at around 4 V vs. Li+/Li.5–7 This was reportedly very difficult, or at least the capacity was highly irreversible. Upon the first net lithiation, a reversible capacity did emerge as Li4+Mn54+O122− ↔ Li4+p+Mn5(4−c)+O122−, where c = p/5 ≥ 0 tracks the degree of transition-metal cation-redox capacity. This part of the capacity at around 3 V vs. Li+/Li was practically seen to be reversible, and the spinel structure could be maintained as long as p ≤ 2.5 (c ≤ 0.5, Li6.5+Mn53.5+O122−), which gives a theoretical capacity of 135.5 mA h g−1. If p was extended to 3 (c = 0.6, Li7+Mn53.4+O122−), the cation-redox theoretical capacity increased to 162.6 mA h g−1, but the cyclability was poor due to the Jahn–Teller distortion induced phase transformation. In addition to a low capacity and low voltage (135.5 mA h g−1 at 3 V), from the stand-point of making a practical Li-matched full cell using the Li4Mn5O12 cathode against a graphite anode, the following challenges must be considered: (a) one must use pre-lithiated graphite or another lithium source to supply additional cyclable lithium for the first net lithiation Li4+Mn54+O122− → Li4+p+Mn5(4−c)+O122−, (b) there is going to be some Mn-ion dissolution in the liquid electrolyte, which aggressively attacks the graphite anode. For these reasons, Li4Mn5O12 is not considered to be an attractive cathode material. Here, however, we will show that by a low-temperature synthesis at 400 °C, we can make a nanostructured Li4Mn5O12-like material with both p,c-contribution (Mn cation redox capacity) and m,a-contribution (O anion redox capacity), giving a total reversible capacity of 212 mA h g−1. Furthermore, by pairing with a pre-lithiated Sn metal anode in a standard carbonate electrolyte, the dissolved Mn-ion's attack on the anode seems to be less of a problem. This enables the stable cycling of a Li-matched full cell based on the Li4Mn5O12-like cathode for more than 100 cycles.
In previous reports, Takada et al. had synthesized a well-crystallized Li4Mn5O12 with a rechargeable capacity of about 135 mA h g−1 at a voltage of 2.5–3.6 V for only 10 cycles.8 Jiang et al. had reported that Li4Mn5O12 prepared by spray-drying could deliver a discharge capacity of 157.8 mA h g−1 at a voltage range of 2.4–3.7 V and maintained 76% capacity retention after 80 cycles.9 Fu et al. found that Li4Mn5O12 with hierarchical porous nano/micro structure had a capacity of 161 mA h g−1 at a voltage range of 2.0–3.5 V and it maintained a capacity of about 89 mA h g−1 after 100 cycles at 0.5C.10 In most studies, the main capacity appears to originate from the p,c-contribution (Mn cation redox capacity) of Li4Mn5O12 + 2.5e− + 2.5Li+ → Li6.5Mn5O12 at a voltage of ∼3 V vs. Li+/Li, and the four initial lithium ions were not active during the charge/discharge process. Moreover, doping strategies were also tried in order to enhance the energy density of Li4Mn5O12, and the operating voltage was increased to 4.7 V, although there was no significant improvement in the capacity.11
Inspired by the recent understanding of reversible oxygen anion-redox reaction in cathode materials, it behooves one to activate the anion-redox contributions and pursue hybrid cation- and anion-redox (HAC) capacities in cathode materials to boost the energy density.5–7 While the reversible O2− ↔ O− (oxo ↔ peroxo) transition has been demonstrated in Li-rich Li1+xM1−xO2 with a layered structure, such as yLi2MnO3·(1 − y)LiMO2 (M = Mn, Ni, Co mixture), there have been very few attempts in spinel Li4Mn5O12, also written as Li(Mn5/3Li1/3)O4, where 1/6 of the Mn ions in the classical LiMn2O4 spinel structure are replaced by Li ions.4,12 Generally speaking, there is a mixture of ionic and covalent bonding between the cations (Li, Mn) and oxygen. While Li–O bonding is strongly ionic, Mn3d/O2p have a significant orbital overlap and thus Mn–O has a stronger covalent bonding character, which stabilizes O2− and pushes Ueq(O1−/2−) to higher voltages. However, the degree of ionic bonding increases with 1/6 of the Mn ions being replaced by Li ions, and as a result, Ueq(O1−/2−) can move to lower voltages, at around 4 V vs. Li+/Li. Thus, the oxygen ions can, in principle, deliver extra m,a-capacities before the voltage gets so high (like >5 V) that the Al current collector corrodes or the electrolyte decomposes too violently.13
Herein, we report a simple and scalable solid-state method to synthesize nanoscale Li4Mn5O12-like particulates at a low temperature. After an activation process, both the cation-redox capacity at ∼3 V vs. Li+/Li originating from Mn3.5+ → Mn4+ and the anion-redox capacity at ∼4 V vs. Li+/Li provided by O2− → O− are obtained. The reaction proceeds theoretically as
| Li2Mn5O12 + 4.5e−(U) + 4.5Li+(electrolyte) ↔ Li6.5Mn5O12 | (1) |
 | ||
| Fig. 1 (a) Hybrid oxygen anion-redox and transition-metal cation-redox reaction coordinates of reaction (1): Li2+Mn54+O121.833− ↔ Li4+Mn54+O122− ↔ Li6.5+Mn53.5+O122−. The inset shows the spinel Li(Mn5/3Li1/3)O4 with a long-range order of 1/6 LiMn substitutions. However, we believe our material may have significant disordering, or a random solid solution, of these 1/6 LiMn substitutions, due to the low-temperature synthesis, (b) Rietveld refinement XRD patterns, (c) Williamson–Hall plots with linear fitting, (d)Mn 2p XPS spectra. | ||
The low-temperature solid-state synthesis process is illustrated in Fig. S1 (ESI†). The Li4Mn5O12-like cathode was prepared through a solid-state process, which is very simple and scalable. The as-obtained sample was then characterized by X-ray diffraction (XRD), and the XRD pattern (Fig. 1b) could be approximately indexed to the Li4Mn5O12 phase (JCPDS no. 46-0810). Based on the Rietveld refinement, the lattice parameters of a monoclinic unit cell are a = 8.128 Å and unit cell volume = 537.130 Å3, closely approaching the values reported by Takada, which confirms its Li4Mn5O12-like motif.14 From the Scherrer equation and Williamson–Hall analysis,15 we found that the particle size was 9.57 nm and the microstrain fluctuation (slope of broadening vs. 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ) was ∼−0.00246 (as shown in Fig. 1c). The small, but negative, value of −0.00246 is unusual and means that the particle sizes are so small that the anisotropic surface stress and other factors give a non-uniform strain distribution. The sub-10 nm nanoparticulate nature (due to the low-T synthesis, which prevented coarsening) is believed to play a key role in the ability to utilize the anion-redox capacity. When Mn stays at 4+ (the red leg of the reaction coordinate in Fig. 1a), the electronic conductivity has to be low due to the lack of transition-metal polaron conduction (unlike the blue leg, where there is an abundant mixture of Mn4+ and Mn3+). Therefore, one relies more on the surface conduction and electron tunneling mechanisms, which favors smaller particles. On the other hand, while the smaller particles have favorable kinetics, they also have more side reactions with the electrolyte, so an optimal particle size should exist.
sinθ) was ∼−0.00246 (as shown in Fig. 1c). The small, but negative, value of −0.00246 is unusual and means that the particle sizes are so small that the anisotropic surface stress and other factors give a non-uniform strain distribution. The sub-10 nm nanoparticulate nature (due to the low-T synthesis, which prevented coarsening) is believed to play a key role in the ability to utilize the anion-redox capacity. When Mn stays at 4+ (the red leg of the reaction coordinate in Fig. 1a), the electronic conductivity has to be low due to the lack of transition-metal polaron conduction (unlike the blue leg, where there is an abundant mixture of Mn4+ and Mn3+). Therefore, one relies more on the surface conduction and electron tunneling mechanisms, which favors smaller particles. On the other hand, while the smaller particles have favorable kinetics, they also have more side reactions with the electrolyte, so an optimal particle size should exist.
X-ray photoelectron spectroscopy (XPS) was carried out (Fig. 1d) to identify the valence state of Mn. The fitting peaks are at 642.58 eV and 654.08 eV, which are the characteristic peaks of Mn4+ 2p3/2 and Mn4+ 2p1/2, respectively.16,17 The microstructure and morphology of the Li4Mn5O12-like powders were also observed through field emission scanning electron microscopy (FE-SEM), as shown in Fig. S2a and S2b (ESI†). Secondary microparticles with a diameter of around 5 µm can be seen (Fig. S2a, ESI†), and a closer observation in Fig. S2b (ESI†) reveals that the secondary particles are self-assembled by the primary nanoparticles. Such a hierarchical microstructure could also be identified by transmission electron microscopy (TEM) (Fig. S3, ESI†), where subunits of ∼20–100 nm agglomerated and formed ∼5 µm particles. It is generally believed that such nano-sized crystallinity could effectively accommodate the strain of Jahn–Teller distortion through the slippage at the domain wall boundaries, and thus is beneficial to the stability of the cathode material.18–20
The Li4Mn5O12-like cathode was then electrochemically examined in coin cells. After an initial charging process Li4+Mn54+O122− → Li4−m+Mn54+O12(2−a)− (that was largely abandoned in conventional samples due to lack of reversibility), the Li4Mn5O12-like cathode seems to be activated and delivers a capacity of 210 mA h g−1 (at 0.1C) in the first discharge, when the net-additional lithium was provided by the counter electrode. In the following 20 cycles, the specific capacity increases gradually and reaches a maximum value of 212 mA h g−1 (a discharge energy density of 668 W h kg−1) at the 20th cycle. This would be impossible without the anion-redox m,a-contributions. As discussed before, since the tetravalent manganese ions cannot be oxidized, the first charge capacity of 82.6 mA h g−1 that we measured must arise from the O-redox reaction, which turns out to be highly reversible in the following cycles. The progress coordinate of reaction (1) contains a directional change as illustrated in Fig. 1a at Li4+Mn54+O122−, with one part given by the red anion-redox and the other part given by the blue cation-redox leg. From the voltage profile, there is a plateau at 2.8 V vs. Li+/Li that corresponds to lithium insertion into the octahedral sites of the spinel Li4Mn5O12 accompanied by the reduction of manganese ions Li4+Mn54+O122− ↔ Li4+p+Mn5(4−c)+O122−. It is commonly believed that Jahn–Teller distortion has no chance of causing a phase change/structural collapse as long as the oxidation state of the manganese ions stays above 3.5.4
Our Li4Mn5O12-like cathode, which was prepared by a simple one-pot synthesis and was not optimized, could still maintain a capacity of 153 mA h g−1 and an energy density of 470 W h kg−1 at room temperature after 145 cycles, as shown in Fig. 2c. Interestingly, the capacity exhibits an increasing trend during the initial 20 cycles, which we believe is associated with an activation process. From the 30th cycle, the capacity starts to decay, possibly originating from the dissolution of manganese with a disproportionation reaction of Mn3+(solid) → Mn4+(solid) + Mn2+(solution) and the onset of Jahn–Teller distortion at the end of discharge.4 The discharge potential of the Li4Mn5O12-like cathode is stable (at 3.1 V) during cycling, almost without any drop (in Fig. 2b).
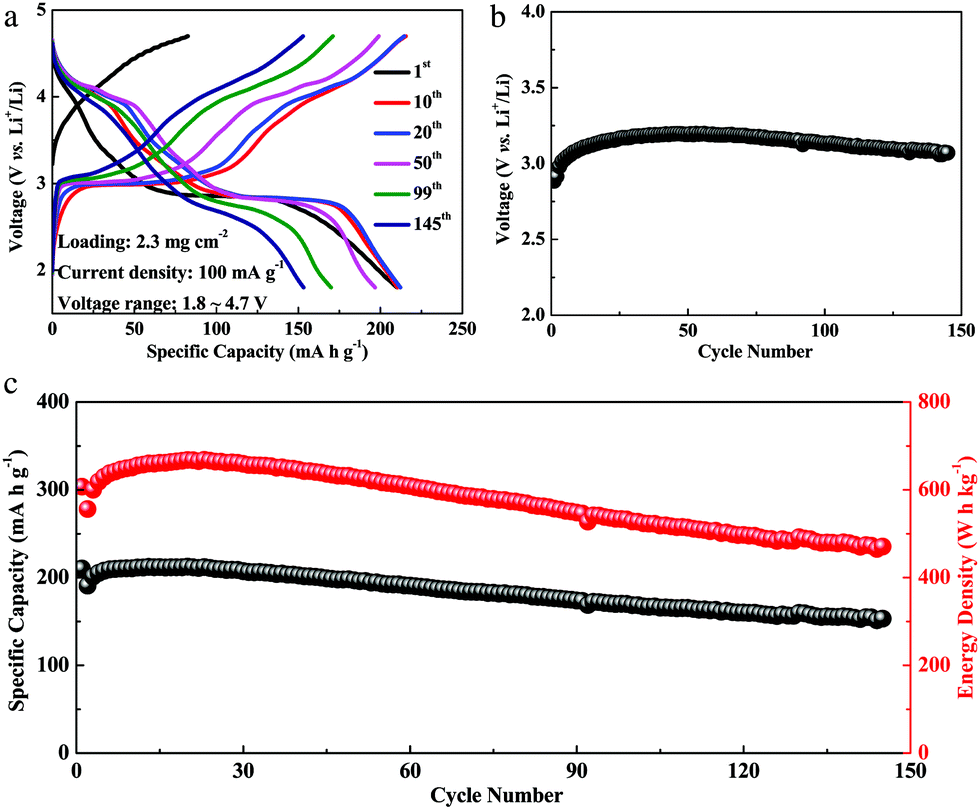 | ||
| Fig. 2 (a) Charge/discharge curves of the Li4Mn5O12-like half cells from the 1st cycle to the 145th cycle at a current density of 100 mA g−1 among 1.8–4.7 V versus Li+/Li at room temperature, (b) evolution of average discharge voltage during cycling, (c) cycling performance of Li4Mn5O12-like half cells. | ||
The full cells were assembled by pairing the Li4Mn5O12-like cathode against a LixSn anode32 and tested at a current density of 100 mA g−1 between 1.0 and 4.1 V at room temperature. As shown in Fig. S6a and S6b (ESI†), the initial discharge capacity reaches 213 mA h g−1. After 45 cycles, the capacity stabilizes at 204 mA h g−1 with hardly any capacity attenuation.
Based on the analysis above, the discharge capacities of the Li4Mn5O12-like cathode include hybrid cation- and anion-redox capacities. Here, we separate the capacities into two parts in a discharge curve of the 20th cycle, which are attributed to the cation-redox and anion-redox processes, as shown in Fig. 3a and b. The cation-redox process (Mn3+/Mn4+) covers the area at about 3 V vs. Li+/Li. The capacity due to this process is 108.4 mA h g−1, corresponding to a reaction of Li4Mn54+O122− + 2Li+ + 2e− ↔ Li6Mn53.6+O122−. The capacity attributed by the anion-redox process is 103.6 mA h g−1, which can be described as a reaction of Li4Mn54+O122− − 1.9Li+ − 1.9e− ↔ Li2.1Mn54+O121.84−. To clarify the underlying mechanism of the anion-redox contribution, we conducted a Differential Electrochemical Mass Spectrometry (DEMS) test under a galvanostatic condition, as shown in Fig. 3c. During the first charging process, O2 was generated once the potential exceeded 3.4 V, implying that there was O2− oxidation. From the second cycle, no O2 generation was detected, indicative of a possible existence of solid-state peroxide, which was further confirmed through XPS characterization (Fig. 3d). From the O 1s spectrum, there is an obvious difference in the oxidation state of the oxygen atoms in the Li4Mn5O12-like cathode at different SOCs. Specifically, for a pristine sample, a peak at 529.5 eV that corresponds to the O2− anions of the crystalline network21,22 and a peak at 532.1 eV that might be associated with a weakly absorbed surface species, such as CO2, were observed. At 4.7 V, two new peaks occur. The peak at 531.1 eV is related to the onset of a new component with a lower electronic density of oxide ions than O2− ions, indicating that O2− ions were oxidized to O−/O2− or under-coordinated oxygen atoms.21 It disappeared when the sample was discharged to 1.8 V. We anticipate that the reversible appearance/disappearance of the 531.1 eV peak during the charge/discharge process reveals the reversibility of the anion-redox reaction. In addition, the other peak at 533.2 eV corresponds to electrolyte oxidation.22,23
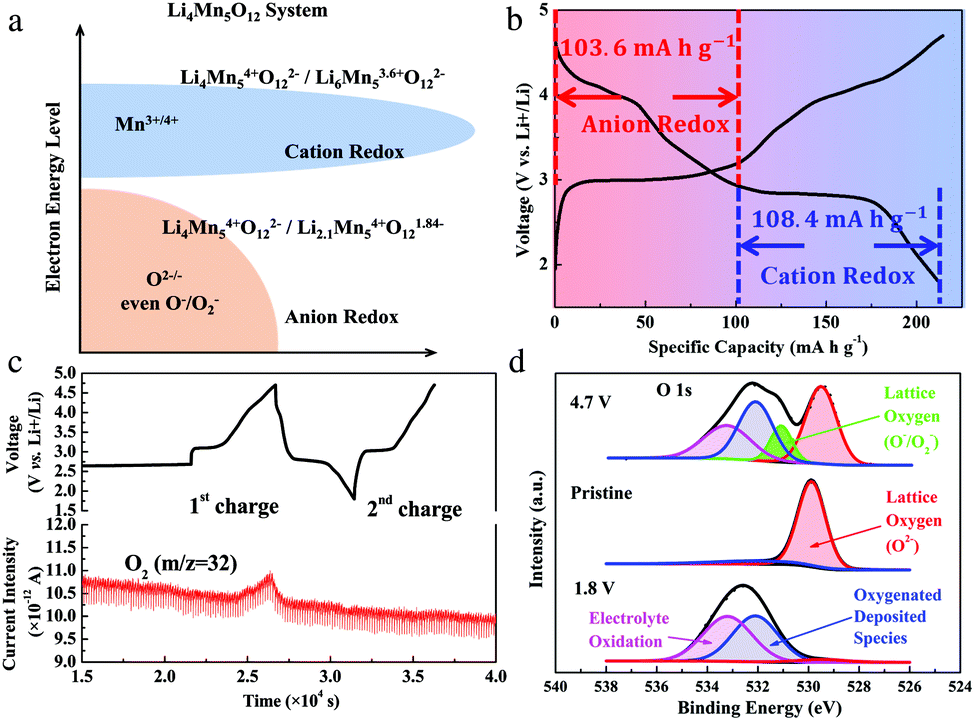 | ||
| Fig. 3 (a) Schematic illustration of cation- and anion-redox processes in the Li4Mn5O12 system, (b) the distinguished cation- and anion-redox capacities in a discharge curve of the 20th cycle respectively, (c) DEMS analysis of the Li4Mn5O12-like cathode in a half cell tested with a constant current density of 100 mA g−1 and a voltage window of 1.8–4.7 V versus Li+/Li, (d) XPS spectra of a pristine Li4Mn5O12-like sample, a sample charged to 4.7 V and a sample discharged to 1.8 V. | ||
We are intrigued as to why our samples manifest a reversible capacity Li4+Mn54+O122− → Li2+Mn54+O121.833− for hundreds of charge/discharge cycles. In addition to the sub-10 nm primary particle size (9.57 nm), we speculate that the partial destruction of chemical ordering could also play a role. Recall that Li4Mn5O12 is better understood as Li(Mn5/3Li1/3)O4, where 1/6 of the Mn sites in the classical LiMn2O4 spinel structure are replaced by Li ions.4,12 However, the Li4Mn5O12 illustrated in Fig. 1a is a perfect compound with chemical ordering, meaning those 1/6 LiMn substitutions are perfectly long-range ordered and not an alloy. However, due to the 400 °C low-T synthesis (most solid-state synthesis takes place at >600 °C),24 the ordering of these LiMn substitutions in our samples may not be perfect, and may form a random solid solution, despite the large agreement in the XRD peaks in Fig. 1b with the reference crystal. That is, while the expression “Li-rich spinel Li(Mn5/3Li1/3)O4” should be correct as a chemical formula, those LiMn substitutions may have more randomized positions. We may also have Mn occupying some of the original tetrahedral Li sites of the classical LiMn2O4 spinel, or other kinds of local ionic disorders. For reference, the Mntetrahedral/octahedral4+ has a Shannon–Prewitt crystal radius of 0.53 Å/0.67 Å, while Litetrahedral/octahedral+ has a Shannon–Prewitt crystal radius of 0.73 Å/0.90 Å.25 This kind of random solid-solution behavior may explain the sloping voltage curve in the first charge in Fig. S6a (ESI†).26–28 Since the detailed Li–O–Mn and Li–O–Li geometries greatly affect the equilibrium oxygen redox potential,29,30 as well as the internal charge-transfer rates (e.g., O1− → O2− and Mn3+ → Mn4+ together), we speculate that this kind of occupancy disorder and random-solution behavior with the redox-activated dynamic rearrangements of ions6 could facilitate an oxygen redox reaction, also assisted, of course, by the small particle size and electron tunneling.
In summary, a cobalt-free Li4Mn5O12-like nanoparticulate cathode, synthesized at a low temperature of 400 °C (most solid-state synthesis of cathode materials occurs at above 600 °C24,31), demonstrates a high reversible capacity of 212 mA h g−1 with hybrid cation- and anion-redox contributions. Compared to commercial LiFePO4, LiCoO2, and Li(NixCoyMn1−x−y)O2, it not only has a lower cost, but also higher capacities (Fig. S9, ESI†). Furthermore, the cycling stability is also acceptable. The Li4Mn5O12-like cathode maintains a capacity of 153 mA h g−1 after 145 cycles at a current density of 100 mA g−1. The electrochemical performance of the full cell with this Li4Mn5O12-like cathode and lithium tin alloy anode is impressive with a high capacity of 214 mA h g−1 and almost without any capacity fading after 45 cycles. Results above show that this Li-rich spinel, also known as Li(Mn5/3Li1/3)O4, is attractive for further investigations as a lithium-ion battery cathode.
Conflicts of interest
There are no conflicts to declare.References
- A. R. Armstrong and P. G. Bruce, Nature, 1996, 381, 499 CrossRef CAS
.
- M. M. Thackeray, A. de Kock, M. H. Rossouw, D. Liles, R. Bittihn and D. Hoge, J. Electrochem. Soc., 1992, 139, 363–366 CrossRef CAS
- E. Ferg, R. Gummow, A. De Kock and M. Thackeray, J. Electrochem. Soc., 1994, 141, L147–L150 CrossRef CAS
- M. M. Thackeray, Prog. Solid State Chem., 1997, 25, 1–71 CrossRef CAS
- Z. Zhu, A. Kushima, Z. Yin, L. Qi, K. Amine, J. Lu and J. Li, Nat. Energy, 2016, 1, 16111 CrossRef CAS
- W. E. Gent, K. Lim, Y. Liang, Q. Li, T. Barnes, S.-J. Ahn, K. H. Stone, M. McIntire, J. Hong and J. H. Song, Nat. Commun., 2017, 8, 2091 CrossRef PubMed
- X. Rong, E. Hu, Y. Lu, F. Meng, C. Zhao, X. Wang, Q. Zhang, X. Yu, L. Gu and Y.-S. Hu, Joule, 2018, 3, 503–517 CrossRef
- T. Takada, H. Hayakawa, E. Akiba, F. Izumi and B. C. Chakoumakos, J. Power Sources, 1997, 68, 613–617 CrossRef CAS
- Y. Jiang, J. Xie, G. Cao and X. Zhao, Electrochim. Acta, 2010, 56, 412–417 CrossRef CAS
- Y. Fu, H. Jiang, Y. Hu, L. Zhang and C. Li, J. Power Sources, 2014, 261, 306–310 CrossRef CAS
- Z. Xie, H. Eikhuemelo, J. Zhao, C. Cain, W. Xu and Y. Wang, J. Electrochem. Soc., 2015, 162, A1523–A1529 CrossRef CAS
- M.-J. Lee, E. Lho, P. Bai, S. Chae, J. Li and J. Cho, Nano Lett., 2017, 17, 3744–3751 CrossRef CAS PubMed
- L. Suo, W. Xue, M. Gobet, S. G. Greenbaum, C. Wang, Y. Chen, W. Yang, Y. Li and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 201712895 Search PubMed
- T. Takada, H. Hayakawa and E. Akiba, J. Solid State Chem., 1995, 115, 420–426 CrossRef CAS
- A. K. Zak, W. A. Majid, M. E. Abrishami and R. Yousefi, Solid State Sci., 2011, 13, 251–256 CrossRef
- V. Di Castro, G. Polzonetti, G. Contini, C. Cozza and B. Paponetti, Surf. Interface Anal., 1990, 16, 571–574 CrossRef CAS
- B. J. Tan, K. J. Klabunde and P. M. A. Sherwood, J. Am. Chem. Soc., 1991, 113, 855–861 CrossRef CAS
- H. Wang, Y. I. Jang and Y. M. Chiang, Electrochem. Solid-State Lett., 1999, 2, 490–493 CrossRef CAS
- Y. I. Jang, B. Huang, Y. M. Chiang and D. R. Sadoway, Electrochem. Solid-State Lett., 1998, 1, 13–16 CrossRef CAS
- Y. Shao-Horn, S. A. Hackney, A. R. Armstrong, P. G. Bruce, R. Gitzendanner, C. S. Johnson and M. M. Thackeray, J. Electrochem. Soc., 1999, 146, 2404–2412 CrossRef CAS
- J.-C. Dupin, D. Gonbeau, P. Vinatier and A. J. P. C. C. P. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC
- M. Sathiya, G. Rousse, K. Ramesha, C. Laisa, H. Vezin, M. T. Sougrati, M.-L. Doublet, D. Foix, D. Gonbeau and W. J. N. m. Walker, Nat. Mater., 2013, 12, 827 CrossRef CAS PubMed
- R. Dedryvere, D. Foix, S. Franger, S. Patoux, L. Daniel and D. Gonbeau, J. Phys. Chem. C, 2010, 114, 10999–11008 CrossRef CAS
- L. Li, Y. Xu, X. Sun, R. Chang, Y. Zhang, X. Zhang and J. Li, Adv. Energy Mater., 2018, 8, 1801064 CrossRef
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef
- J. Niu, A. Kushima, X. Qian, L. Qi, K. Xiang, Y.-M. Chiang and J. Li, Nano Lett., 2014, 14, 4005–4010 CrossRef CAS PubMed
- H. Liu, F. C. Strobridge, O. J. Borkiewicz, K. M. Wiaderek, K. W. Chapman, P. J. Chupas and C. P. Grey, Science, 2014, 344, 1252817 CrossRef PubMed
- M. Z. Bazant, Acc. Chem. Res., 2013, 46, 1144–1160 CrossRef CAS PubMed
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef CAS PubMed
- A. Urban, I. Matts, A. Abdellahi and G. Ceder, Adv. Energy Mater., 2016, 6, 1600488 CrossRef
- J. L. Shi, D. D. Xiao, M. Ge, X. Yu, Y. Chu, X. Huang, X. D. Zhang, Y. X. Yin, X. Q. Yang and Y. G. Guo, Adv. Mater., 2018, 30, 1705575 CrossRef PubMed
- H. Xu, S. Li, C. Zhang, X. L. Chen, W. J. Liu, Y. H. Zheng, Y. Xie, Y. H. Huang and J. Li, Energy Environ. Sci., 2019 10.1039/C9EE01404G
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc02006c |
| This journal is © The Royal Society of Chemistry 2019 |