
Photoactive Zn–air batteries using spinel-type cobalt oxide as a bifunctional photocatalyst at the air cathode†

Abstract
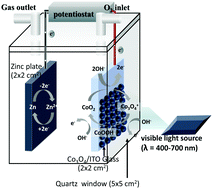
Spinel-type cobalt oxide (Co3O4) was synthesized and used as a photoactive bifunctional electrocatalyst towards the oxygen evolution reaction (OER) and the oxygen reduction reaction (ORR) at the air cathode of zinc–air batteries (ZABs). The Co3O4 having direct and indirect band gap energies of ca. 2.20 eV and ca. 1.35 eV can absorb visible light, generating photogenerated carriers and photoelectrons via the photoelectric effect. Upon exposure to visible light, the Co3O4 electrode exhibits ca. 30% higher current density than that under dark conditions and provides around 10–20% lower OER and ORR overpotentials than those under the dark conditions. Under visible light, the specific capacity of the as-fabricated photoactive ZAB cell is improved by ca. 10% as compared to that under dark conditions.
 Please wait while we load your content...
Please wait while we load your content...

