
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of cAAC stabilized biradical of “Me2Si” and “Me2SiCl” monoradical from Me2SiCl2 – an important feedstock material†
Soumen
Sinhababu
 a,
Subrata
Kundu
a,
Mujahuddin M.
Siddiqui
a,
Alexander N.
Paesch
a,
Regine
Herbst-Irmer
a,
Brigitte
Schwederski
b,
Pinaki
Saha
c,
Lili
Zhao
*c,
Gernot
Frenking
*cd,
Wolfgang
Kaim
*b,
Dietmar
Stalke
*a and
Herbert W.
Roesky
*a
a,
Subrata
Kundu
a,
Mujahuddin M.
Siddiqui
a,
Alexander N.
Paesch
a,
Regine
Herbst-Irmer
a,
Brigitte
Schwederski
b,
Pinaki
Saha
c,
Lili
Zhao
*c,
Gernot
Frenking
*cd,
Wolfgang
Kaim
*b,
Dietmar
Stalke
*a and
Herbert W.
Roesky
*a
aInstitut für Anorganische Chemie, Universität Göttingen, Tammannstrasse 4, 37077 Göttingen, Germany. E-mail: hroesky@gwdg.de; dstalke@chemie.uni-goettingen.de
bUniversität Stuttgart, Institut für Anorganische Chemie, 70569 Stuttgart, Germany. E-mail: kaim@iac.uni-stuttgart.de
cInstitute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing Tech University, Nanjing 211816, China
dUniversität Marburg, Fachbereich Chemie, Hans-Meerwein-Strasse, 35032 Marburg, Germany
First published on 20th March 2019
Abstract
The cyclic alkyl(amino) carbene (cAAC) coordinated biradical of dimethylsilicon was isolated as (cAAC)2Me2Si (1), (cAAC = C(CH2)(CMe2)2N-2,6-i-Pr2C6H3), synthesized from the reduction of Me2SiCl2 using two equivalents of KC8 in the presence of two equivalents of cAAC. The reduction of Me2SiCl2 by one equivalent of KC8 in the presence of one equivalent of cAAC resulted in the stable dimethylsiliconchloride monoradical (cAAC)Me2SiCl (2).
Radicals and biradicals have attracted considerable attention in chemistry and material science due to their unique optical, magnetic and electronic properties.1 In 1915 Schlenk isolated the first paramagnetic biradical from the reaction of bis-diphenyl-benzyl dichloride with a copper–tin alloy.2a Most of the radicals are unstable and are short-lived.2b–d However, they can be isolated and stored at room temperature in a pure form using either thermodynamic or kinetic stabilization.3,4 Several stable carbon and silicon centered biradicals are known.5 Carbon centered 1,3-biradicals are proposed to be key reactive intermediates in certain chemical reactions.6 Limited examples of four-membered heterocyclic 1,3-biradicals of cyclobutane type were reported.7 Recently, we have synthesized air stable carbon centered 1,3-biradical (cAAC)2SiCl2 (I)8 (Chart 1) from the reaction of NHC → SiCl2 (NHC = N-heterocyclic carbene) with a cyclic alkyl(amino) carbene. Furthermore, our group successfully synthesized cAAC stabilized SiX2 (X = H, F) (Chart 1) bridged 1,3-biradicals, which are stable at room temperature for more than three months under inert atmosphere. The elusive SiF2 bridged biradical (cAAC)2SiF2 (III) (Chart 1) was synthesized from the reduction of (cAAC)SiF4 by using two equivalents of KC8 in the presence of one equivalent of cAAC.9 While, (cAAC)2SiH2 (IV) (Chart 1) was prepared from the reduction of H2SiI2 with two equivalents of KC8, in the presence of two equivalents of cAAC.10 After the successful isolation of cAAC stabilized SiX2 (X = H, Cl, F) bridged 1,3-biradicals, the isolation of SiMe2 analogues (cAAC)2Me2Si (1) was a prominent missing link in this class of compounds. Dimethyl silicon is not stable at room temperature and polymerises to (SiMe2)n. In our earlier synthetic route, we tried to isolate 1, by the reaction of (cAAC)2SiCl2 with 2 equivalents of MeLi by the nucleophilic substitution method.11 To our surprise, MeLi functioned as a reducing agent leading to the isolation of dehalogenated biradicaloid (cAAC)2Si (II).12 Me2SiCl2 is the most important feedstock material in the industry for the preparation of silicones.13 We envisaged an alternative route to the isolation of 1 by the reduction of commercially available Me2SiCl2. Herein, we report a one step synthesis of the biradical (cAAC)2Me2Si (1) and monoradical (cAAC)Me2SiCl (2) by the reduction of Me2SiCl2 with KC8.
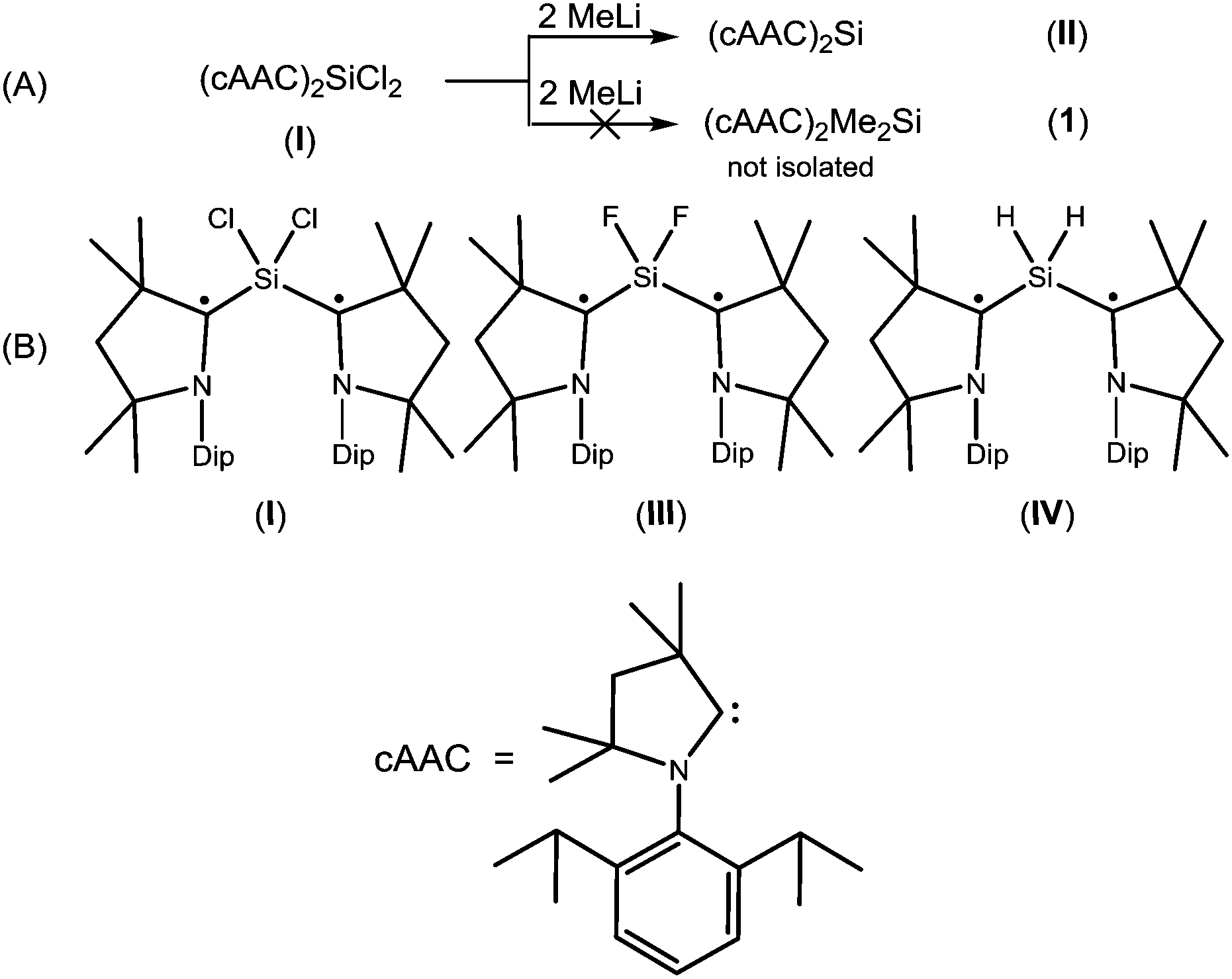 | ||
| Chart 1 (A) The unsuccessful attempted synthesis of (cAAC)2Me2Si (1); (B) structurally characterized stable biradicals containing SiX2 (X = Cl, F, H) moiety8–10 | ||
Both the compounds 1 and 2 were fully characterized by X-ray crystallography and EPR spectroscopy. Compound 1 was prepared by reduction of Me2SiCl2 using KC8 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio in the presence of two equivalents of cAAC (Scheme 1; for details, see ESI†). 1H NMR spectrum of compound 1 shows broad resonance indicating the radical nature. 1 has been characterized by EPR spectroscopy, LIFDI mass spectrometry, elemental analysis and single crystal structure analysis. 1 is stable in an inert atmosphere for more than 6 months in the solid state. It is thermally stable at room temperature and decomposes at 148 °C. The UV/Vis spectrum of 1 in a hexane solution shows an absorption band at 575 nm. The LIFDI mass spectrum in toluene exhibits a peak at 629.6 m/z for [M]+. Single crystals of 1 suitable for X-ray diffraction analysis were grown from hexane solution at −26 °C.
:2 molar ratio in the presence of two equivalents of cAAC (Scheme 1; for details, see ESI†). 1H NMR spectrum of compound 1 shows broad resonance indicating the radical nature. 1 has been characterized by EPR spectroscopy, LIFDI mass spectrometry, elemental analysis and single crystal structure analysis. 1 is stable in an inert atmosphere for more than 6 months in the solid state. It is thermally stable at room temperature and decomposes at 148 °C. The UV/Vis spectrum of 1 in a hexane solution shows an absorption band at 575 nm. The LIFDI mass spectrum in toluene exhibits a peak at 629.6 m/z for [M]+. Single crystals of 1 suitable for X-ray diffraction analysis were grown from hexane solution at −26 °C.
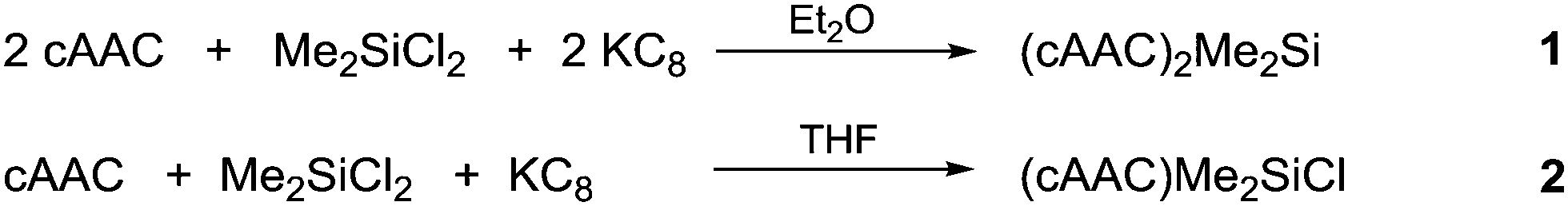 | ||
| Scheme 1 Synthesis of 1 and 2. | ||
A stable radical containing the Me2SiCl group has not been reported so far. Equivalent amounts of cAAC, Me2SiCl2 and KC8, respectively, treated in THF at −90 °C resulted in the desired monoradical product (cAAC)Me2SiCl (2) as orange coloured crystals in 68% yield (Scheme 1).
2 was characterized by EPR spectroscopy, LIFDI mass spectrometry, elemental analysis and single crystal structure analysis. The LIFDI mass spectrum in toluene exhibits a molecular ion peak at 378.2 m/z. The UV/Vis spectrum of 2 in hexane shows an absorption band at 435 nm. The EPR spectra of compounds 1 and 2 were recorded in hexane solution at room temperature. It must be mentioned that efforts to isolate such type of radical species with NHC were not successful.
1 crystallizes in the monoclinic space group C2/c. The molecular structure of 1 (Fig. 1) reveals the central Si atom to be distorted tetrahedrally coordinated by four carbon atoms. The Si–Me bond lengths [1.8768(13) and 1.8800(13) Å] are similar to the CcAAC–Si distances [1.8814(13) and 1.8829(13) Å] and to the distances reported in the literature.10,14 The C2–Si1–C2A bond angle (116.86(5)°) is widened due to the steric hindrance of the bulky cAAC ligands, but nevertheless smaller than in the silylone (cAAC)2Si of 119.10(1).12b
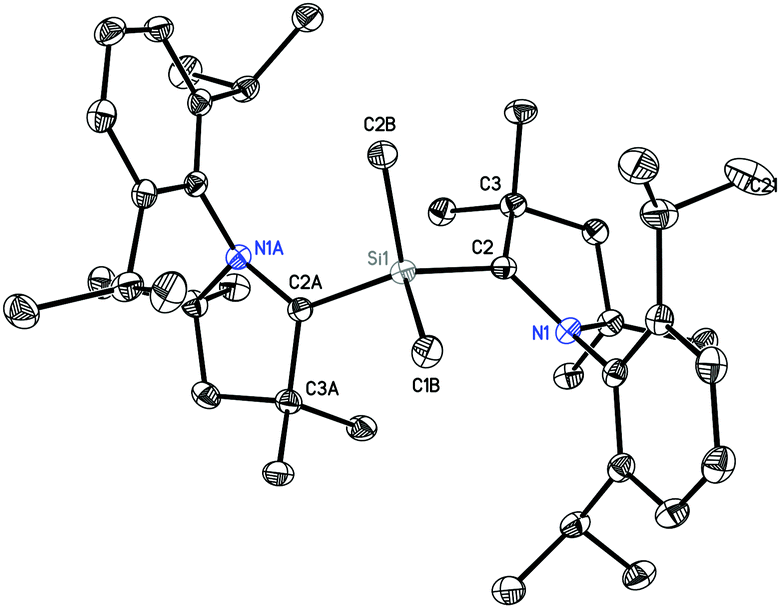 | ||
| Fig. 1 Crystal structure of 1. Hydrogen atoms are omitted for clarity. Thermal ellipsoid plot is drawn at 50% probability. Selected experimental bond lengths [Å] and angles [°]. Calculated values at BP86/def2-TZVP are given in brackets: Si1–C2B, 1.8768(13) [1.891], Si1–C1B, 1.8800(13) [1.891]; Si1–C2; 1.8814(13) [1.886]; Si1–C2A, 1.8829(13) [1.886]; C2B–Si1–C1B, 106.55(6) [106.6]; C2B–Si1–C2, 108.33(6) [108.7]; C1B–Si1–C2, 107.98(6) [108.9]; C2B–Si1–C2A, 108.40(6) [108.7]; C1B–Si1–C2A, 108.27(6) [108.7]; C2–Si1–C2A, 116.86(5) [114.9]. | ||
2 crystallizes in the orthorhombic space group Pbca. The molecular structure (Fig. 2) reveals the silicon atom to be tetra-coordinated with three carbon and one chlorine atom. The Si–Cl bond length (2.1228(5) Å) is longer than those in (MecAAC)SiCl3 (2.0396(4)–2.0864(3) Å).15
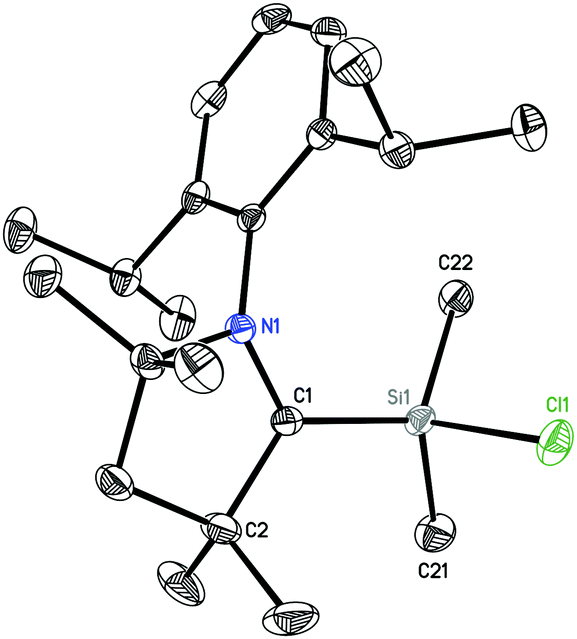 | ||
| Fig. 2 Crystal structure of 2. Hydrogen atoms are omitted for clarity. Thermal ellipsoid plot is drawn at 50% probability. Selected experimental bond lengths [Å] and angles [°]. Calculated values at BP86/def2-TZVP are given in brackets: Si1–C1, 1.8323(12) [1.841]; Si1–C22, 1.8633(13) [1.878]; Si1–C21, 1.8645(13) [1.876]; Si1–Cl1, 2.1228(5) [2.138]; C1–Si1–C22, 117.67(6); C1–Si1–C21, 113.16(6); C22–Si1–C21, 108.22(6); C1–Si1–Cl1, 108.56(4) [108.1]; C22–Si1–Cl1, 104.05(4) [104.5]; C21–Si1–Cl1, 103.91(5) [105.0]. | ||
The EPR spectrum of 1 is dominated by a 1:1:1 triplet of 4.65 G (Fig. 3), attributed to the coupling of the unpaired electron with one 14N atom (I = 1). This splitting9,10,15 suggests localized spin at only one of the two equivalent cAAC groups connected to the silicon atom which exhibits a typically 29Si isotope coupling9,10,15 of 27.5 G (29Si: I = 1/2, 4.7% nat. abundance).
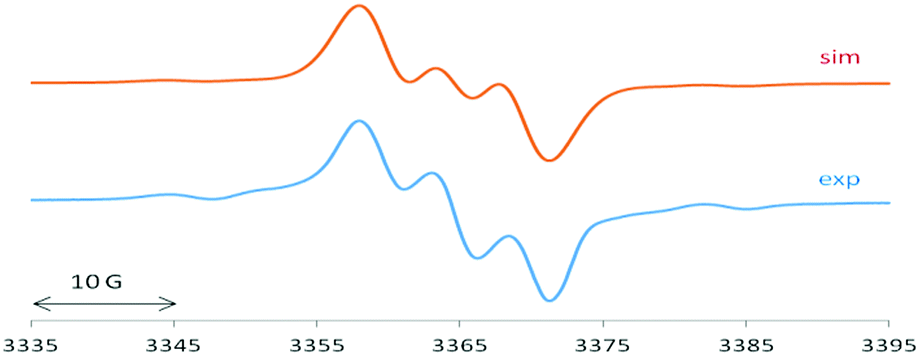 | ||
| Fig. 3 EPR spectrum of 1 with computer simulation (top). For parameters see main text. | ||
2 exhibits an EPR spectrum (Fig. 4) with similar 14N and 29Si values of 5.4 G and 25 G, respectively, in addition to a sizeable chlorine splitting from the isotopes 35Cl (I = 3/2, 75.8% nat. abundance: 8.9 G) and 37Cl (I = 3/2, 24.2%: 7.4 G). Such Cl(Si) coupling has been noted before for related silicon radicals.15
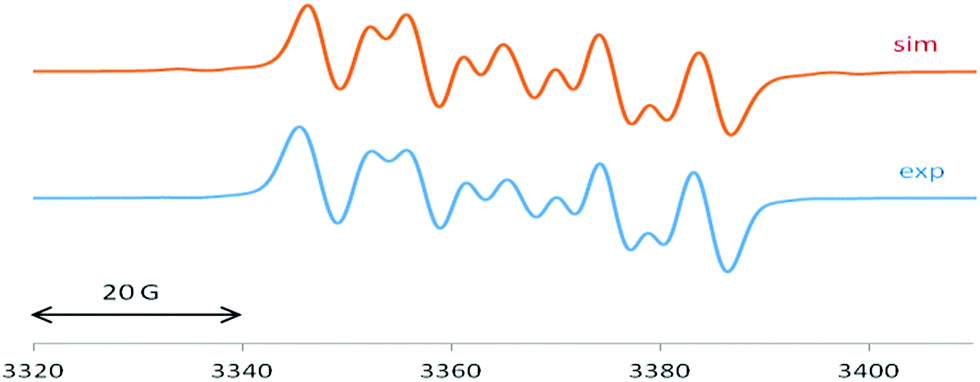 | ||
| Fig. 4 EPR spectrum of 2 with computer simulation (top). For parameters see main text. | ||
We carried out quantum chemical calculations using density functional theory at the BP86/def2-TZVP level23 in order to analyze the electronic structure of compounds 1 and 2. Fig. 1 and 2 shows also the computed bond lengths and angles of the optimized geometries of the two molecules, which are in excellent agreement with the experimental data. The calculations suggest that 1 has an electronic triplet ground state whereas 2 is a doublet, which concurs with the EPR results. Fig. 5 shows the spin density distribution of the two molecules. The unpaired electrons in 1 and 2 are mainly located at the nitrogen atoms and the carbene carbon atoms of the cAAC moieties.
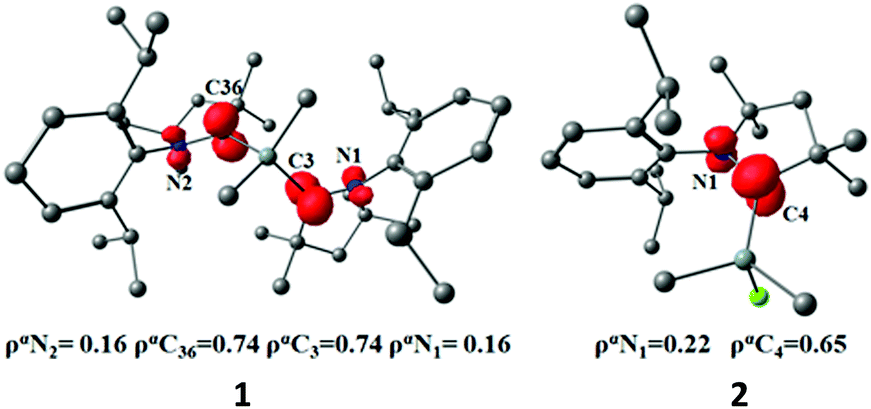 | ||
| Fig. 5 Spin density at the of compounds 1 and 2 at the BP86/def2-TZVP level. | ||
We further analyzed the nature of the cAAC–Si bonds in 1 and 2 with the EDA-NOCV method.24 Table S1 (ESI†) shows the numerical results. The calculations for 1 were carried out using the cAAC fragments in the electronic triplet state, which gives an overall quintet state for the (cAAC)2 ligand, and the SiMe2 moiety in the triplet state. For compound 2 we took the cAAC ligand in the triplet state and the SiMe2Cl fragment in the doublet state. The choice of the open-shell fragments corresponds to electron-sharing single bonds. Comparative calculations using an electronic singlet state spin for the cAAC ligands, which correspond to dative bonds cAAC → Si, gave significantly larger orbital values ΔEorb (see Tables S2 and S3 in ESI†). It has been shown in previous studies that the orbital values ΔEorb are a probe for the choice of the best fragments.25
The data in Table S1 (ESI†) show that the covalent orbital interactions ΔEorb have nearly equal strength as the Coulomb attraction. There are two major orbital contributions ΔEorb(1) and ΔEorb(2) in compound 1 and one dominant term ΔEorb(1) in 2, which come from pairwise orbital interactions between the chosen fragments. Fig. S5 (ESI†) shows the plots of the associated deformation densities Δρ, which illustrate the charge flow that is connected to the orbital interactions. The color code of the charge flow indicates the direction red → blue. The complete list of the deformation densities Δρ and the connected fragment orbitals are shown in Fig. S3 and S4 of ESI.† It becomes obvious that ΔEorb(1) in compound 1 is due to the interaction between the singly occupied σ orbital (SOMO) of SiMe2 with the in-phase (+, +) combination of the π-type26 SOMO of (cAAC)2, where the net charge flow is from SiMe2 → (cAAC)2. The stabilization energy ΔEorb(2) in compound 1 comes from the interaction of the π SOMO of SiMe2 with the out-of-phase (+, −) combination of the σ-type26 SOMO of (cAAC)2. The dominant orbital interaction ΔEorb(1) in compound 2 is due to the interaction between the σ SOMO of SiClMe2 with the σ SOMO of cAAC. The direction of the charge flow between the ligands is in agreement with the calculated partial charges by the NBO27 method. The computed charges q at the BP86/def2-TZVP level are q(SiMe2) = +0.82 e for 1 and q(SiClMe2) = +0.36 e for 2. Thus, the cAAC ligand in 1 and 2 acts as an acceptor rather than donor.
In summary, we report on the synthesis of 1,3-biradical containing Me2Si moiety using cAAC as ligand. Moreover we isolated the monoradical (cAAC)Me2SiCl. Theoretical investigations and EPR spectra of both compounds have been reported. The calculations suggest that 1 has an electronic triplet ground state whereas 2 is a doublet.
Crystal structure determination. Single crystals were selected and covered with perfluorinated polyether oil on a microscope slide.16 An appropriate crystal was selected using a polarize microscope, mounted on the tip of a MiTeGen©MicroMount, fixed to a goniometer head and shock cooled by the crystal cooling device. The data of 1 and 2 were collected from shock-cooled crystals at 100(2) K on a BRUKER D8 three circle diffractometer equipped with an INCOATEC Mo microsource with mirror optics (MoKα radiation, λ = 0.71073 Å) and smart APEX II detector. They were integrated with SAINT.17 A multi-scan absorption correction and a 3λ correction18 was applied using SADABS.19 The structure were solved by direct methods (SHELXT)20 and refined by full-matrix least-squares methods against F2 (SHELXL)21 in the graphical user interface ShelXle.22 CCDC 1894458 (1) and 1894459 (2).†
H. W. R. is thankful to the DFG for financial support (RO 224/68-1). D. S. thanks the Danish National Research Foundation (DNRF93) funded Centre for Materials Crystallography (CMC) for partial support. We thank Dr A. C. Stückl for EPR measurements. G. F. and L. Z. thank Nanjing Tech University (Grant No. 39837132 and 39837123) and SICAM Fellowship from Jiangsu National Synergetic Innovation Center for Advanced Materials. L. Z. acknowledges the financial support from National Natural Science Foundation of China (Grant No. 21703099), Natural Science Foundation of Jiangsu Province (Grant No. BK20170964), and the high performance center of Nanjing Tech University for supporting the computational resources.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed; (b) Y. Morita, S. Nishida, T. Murata, M. Moriguchi, A. Ueda, M. Satoh, K. Arifuku, K. Sato and T. Takui, Nat. Mater., 2011, 10, 947–951 CrossRef CAS PubMed; (c) I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC; (d) Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- (a) W. Schlenk and M. Brauns, Chem. Ber., 1915, 48, 661–669 CrossRef CAS; (b) G. Porter and M. W. Windsor, Nature, 1957, 180, 187–188 CrossRef CAS; (c) E. G. Janzen and B. J. Blackburn, J. Am. Chem. Soc., 1968, 90, 5909–5910 CrossRef CAS; (d) M. Abe, W. Adam and W. M. Nau, J. Am. Chem. Soc., 1998, 120, 11304–11310 CrossRef CAS.
- Organic and organometallic monoradicals: (a) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 15620–15623 CrossRef CAS PubMed; (b) D. Mandal, R. Dolai, N. Chrysochos, P. Kalita, R. Kumar, D. Dhara, A. Maiti, R. S. Narayanan, G. Rajaraman, C. Schulzke, V. Chandrasekhar and A. Jana, Org. Lett., 2017, 19, 5605–5608 CrossRef CAS PubMed; (c) D. Rottschäfer, B. Neumann, H.-G. Stammler, M. van Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 4765–4768 CrossRef PubMed; (d) S. Styra, M. Melaimi, C. E. Moore, A. L. Rheingold, T. Augenstein, F. Breher and G. Bertrand, Chem. – Eur. J., 2015, 21, 8441–8446 CrossRef CAS PubMed; (e) S. Kundu, S. Sinhababu, S. Dutta, T. Mondal, D. Koley, B. Dittrich, B. Schwederski, W. Kaim, A. C. Stückl and H. W. Roesky, Chem. Commun., 2017, 53, 10516–10519 RSC; (f) M. M. Siddiqui, S. K. Sarkar, S. Sinhababu, P. N. Ruth, R. Herbst-Irmer, D. Stalke, M. Ghosh, M. Fu, L. Zhao, D. Casanova, G. Frenking, B. Schwederski, W. Kaim and H. W. Roesky, J. Am. Chem. Soc., 2019, 141, 1908–1912 CrossRef CAS PubMed.
- Organic and organometallic biradicals: (a) M. M. Hansmann, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2546–2554 CrossRef CAS PubMed; (b) D. Rottschäfer, N. K. T. Ho, B. Neumann, H.-G. Stammler, M. van Gastel, D. M. Andrada and R. S. Ghadwal, Angew. Chem., Int. Ed., 2018, 57, 5838–5842 CrossRef PubMed; (c) D. Rottschäfer, B. Neumann, H.-G. Stammler, D. M. Andrada and R. S. Ghadwal, Chem. Sci., 2018, 9, 4970–4976 RSC; (d) K. C. Mondal, B. Dittrich, B. Maity, D. Koley and H. W. Roesky, J. Am. Chem. Soc., 2014, 136, 9568–9571 CrossRef CAS PubMed; (e) T. Nozawa, M. Nagata, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2011, 133, 5773–5775 CrossRef CAS PubMed.
- M. Abe, J. Ye and M. Mishima, Chem. Soc. Rev., 2012, 41, 3808–3820 RSC.
- (a) W. G. Bentrude, S.-G. Lee, K. Akutagawa, W.-Z. Ye and Y. Charbonnel, J. Am. Chem. Soc., 1987, 109, 1577–1579 CrossRef CAS; (b) W. Adam, G. Reinhard, H. Platsch and J. Wirz, J. Am. Chem. Soc., 1990, 112, 4570–4571 CrossRef CAS; (c) M. Sakamoto, H. Kawanishi, T. Mino and T. Fujita, Chem. Commun., 2008, 2132–2133 RSC.
- (a) J. Bresien, A. Hinz, A. Schulz and A. Villinger, Dalton Trans., 2018, 47, 4433–4436 RSC; (b) K. Takeuchi, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2011, 133, 12478–12481 CrossRef CAS PubMed; (c) H. Cox, P. B. Hitchcock, M. F. Lappert and L. J.-M. Pierssens, Angew. Chem., Int. Ed., 2004, 43, 4500–4504 CrossRef CAS PubMed; (d) C. Cui, M. Brynda, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2004, 126, 6510–6511 CrossRef CAS PubMed; (e) D. Scheschkewitz, H. Amii, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Science, 2002, 295, 1880–1881 CrossRef CAS PubMed; (f) T. Baumgartner, D. Gudat, M. Nieger, E. Niecke and T. J. Schiffer, J. Am. Chem. Soc., 1999, 121, 5953–5960 CrossRef CAS; (g) E. Niecke, A. Fuchs, F. Baumeister, M. Nieger and W. W. Schoeller, Angew. Chem., Int. Ed. Engl., 1995, 34, 555–557 CrossRef CAS.
- K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, I. Tkach, H. Wolf, D. Kratzert, R. Herbst-Irmer, B. Niepötter and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 1801–1805 CrossRef CAS PubMed.
- S. Sinhababu, S. Kundu, A. N. Paesch, R. Herbst-Irmer, D. Stalke, I. Fernandez, G. Frenking, A. C. Stückl, B. Schwederski, W. Kaim and H. W. Roesky, Chem. – Eur. J., 2018, 24, 1264–1268 CrossRef CAS PubMed.
- S. Kundu, P. P. Samuel, S. Sinhababu, A. V. Luebben, B. Dittrich, D. M. Andrada, G. Frenking, A. C. Stückl, B. Schwederski, A. Paretzki, W. Kaim and H. W. Roesky, J. Am. Chem. Soc., 2017, 139, 11028–11031 CrossRef CAS PubMed.
- K. C. Mondal, P. P. Samuel, M. Tretiakov, A. P. Singh, H. W. Roesky, A. C. Stückl, B. Niepötter, E. Carl, H. Wolf, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2013, 52, 4736–4743 CrossRef CAS PubMed.
- (a) K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, B. Niepötter, H. Wolf, R. Herbst-Irmer and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 2963–2967 CrossRef CAS PubMed; (b) B. Niepötter, R. Herbst-Irmer, D. Kratzert, P. P. Samuel, K. C. Mondal, H. W. Roesky, P. Jerabek, G. Frenking and D. Stalke, Angew. Chem., Int. Ed., 2014, 53, 2766–2770 CrossRef PubMed.
- H.-H. Moretto, M. Schulze and G. Wagner, Silicones, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, pp. 675–712 Search PubMed.
- (a) S. Kundu, S. Sinhababu, M. M. Siddiqui, A. V. Luebben, B. Dittrich, T. Yang, G. Frenking and H. W. Roesky, J. Am. Chem. Soc., 2018, 140, 9409–9412 CrossRef CAS PubMed; (b) M. M. Siddiqui, S. Sinhababu, S. Dutta, S. Kundu, P. N. Ruth, A. Münch, R. Herbst-Irmer, D. Stalke, D. Koley and H. W. Roesky, Angew. Chem., Int. Ed., 2018, 57, 11776–11780 CrossRef CAS PubMed.
- S. Roy, A. C. Stückl, S. Demeshko, B. Dittrich, J. Meyer, B. Maity, D. Koley, B. Schwederski, W. Kaimb and H. W. Roesky, J. Am. Chem. Soc., 2015, 137, 4670–4673 CrossRef CAS PubMed.
- (a) T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615–619 CrossRef; (b) T. Kottke, R. J. Lagow and D. Stalke, J. Appl. Crystallogr., 1996, 29, 465–468 CrossRef CAS; (c) D. Stalke, Chem. Soc. Rev., 1998, 27, 171–178 RSC; (d) see the video at http://www.stalke.chemie.uni-goettingen.de/virtuelles_labor/special/22_de.html.
- SAINT v8.30C in BRUKER APEX II, BRUKER AXS Inst. Inc., Madison, USA, 2014 Search PubMed.
- L. Krause, R. Herbst-Irmer and D. Stalke, J. Appl. Crystallogr., 2015, 48, 1907–1913 CrossRef CAS.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- The details of the theoretical methods are described in the ESI†.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962–975 CrossRef CAS PubMed.
- Recent representative examples: (a) C. Mohapatra, S. Kundu, A. N. Paesch, R. Herbst-Irmer, D. Stalke, D. M. Andrada, G. Frenking and H. W. Roesky, J. Am. Chem. Soc., 2016, 138, 10429–10432 CrossRef CAS PubMed; (b) D. M. Andrada, J. L. Casalz-Sainz, A. M. Pendas and G. Frenking, Chem. – Eur. J., 2018, 24, 9083–9089 CrossRef CAS PubMed; (c) L. Zhao, M. Hermann, N. Holzmann and G. Frenking, Coord. Chem. Rev., 2017, 344, 163–204 CrossRef CAS.
- The symmetry assignments σ and π refer to he local symmetry of the fragments.
- C. R. Landis and F. Weinhold, Valency and Bonding: A Natural Bond Orbital Donor–Acceptor Perspective, Cambridge University Press, Cambridge, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Including experimental section, computational details and details of X-ray structural analysis. CCDC 1894458 (1) and 1894459 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc01448a |
| This journal is © The Royal Society of Chemistry 2019 |