
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSecond generation DNA-encoded dynamic combinatorial chemical libraries†
Francesco V.
Reddavide‡
 *ab,
Meiying
Cui‡
a,
Weilin
Lin
a,
Naiqiang
Fu
a,
Stephan
Heiden
b,
Helena
Andrade
a,
Michael
Thompson
b and
Yixin
Zhang
*a
*ab,
Meiying
Cui‡
a,
Weilin
Lin
a,
Naiqiang
Fu
a,
Stephan
Heiden
b,
Helena
Andrade
a,
Michael
Thompson
b and
Yixin
Zhang
*a
aB CUBE Center for Molecular Bioengineering, Technische Universität Dresden, Germany. E-mail: yixin.zhang1@tu-dreden.de
bDyNAbind GmbH, Dresden, Germany. E-mail: francesco.reddavide@dynabind.com
First published on 1st March 2019
Abstract
We present a DNA-encoded chemical library, which allows dynamic selection followed by ligation of the encoding strands. As a chemical approach to mimic the genetic recombination process of adaptive immunity, the technology led to an enhanced enrichment factor and signal-to-noise ratio compared to static libraries.
As the number and complexity of drug targets increase, we increasingly look for novel technologies to discover quality hits across the chemical space.1–3 In the 1980s, hybridoma technology4 and phage display technology5 were popular for their abilities to present massive numbers of different antibody molecules for drug discovery.6 Until recently, however, chemical libraries were orders of magnitude smaller, largely due to the time and logistical requirements of synthesis and high-throughput screening methodologies.7 One way to fill this gap is by using the DNA-Encoded Chemical Library (DECL) technology, a powerful tool for rapidly selecting binders from millions to billions of diverse, combinatorially-generated organic molecules.8–11 The power of DECL largely relies on the combinatorial synthesis and selection process, which can to some degree mimic the combinatorial mechanism of biology.
Over the last two decades, many advances in DECL aim to mimic the genetic mechanisms of immunity and evolution, including combinatorial assembly,12–14 dynamic recombination,15,16 and adaption.17 One seminal work was the development of Encoded Self-Assembling Combinatorial (ESAC) libraries.13 In such a system, complementary DNA strands are employed to simultaneously present two spatially constrained potential drug fragments to the target, an elegant method to increase the chemical space in de novo drug discovery,18 or to design a biased library based on a known binder.19 Moreover, the ESAC technology reduces the need for split-and-pool synthesis, allowing HPLC purification and quality control for sub-library members.13 However, the strength of ESAC libraries is limited by the fact that the identities of fragments are revealed, but not the pairing information between them. One elegant solution to this problem has been recently presented by using a Klenow fill-in to transfer one code to the other.18
The chemical analogue of dynamic recombination has been realized in the DNA-encoded dynamic combinatorial chemical library (EDCCL).15 As a variation of a dynamic chemical library,20 EDCCL was designed as intrinsically unstable DNA duplexes, in order to combat the problem of low signal-to-noise ratios and high incidences of false positives associated with most of the large libraries.21,22 In the absence of a target protein binding, randomly paired encoded chemical moieties undergo constant reshuffling in solution. Upon binding to the target, the specific pairs can be stabilized. Over time, the process drives thermodynamic equilibrium to the formation of potent fragment pairs, resulting in fewer but more reliable hits. Herein we present a Y-shaped DNA architecture for the EDCCL (Y-EDCCL) design. This geometry allows the dynamic enrichment of potent binding pairs and subsequent ligation to recode the relationships between fragments. Upon allowing both code joining and dynamic features, it would lead to a further step to mimic immunity to generate and evolve small-molecule binders.
Y-EDCCL consists of four DNA strands (Fig. 1). Strands A and B are assembled to form the sub-library Y-5. Strand A is linked to a small molecule on its 5′ end. Strand B is complementary and stably annealed to strand A, on and around the coding region. Strand A has a 5′ overhang of 11 or 13 nucleotides. Similarly, Strands C and D make up the sub-library Y-3 (Fig. 1). Strand C is linked to a small molecule on its 3′ end. Strand D is complementary and stably annealed to Strand C, on and around the coding region. Y-3 has two overhangs, a 3′ overhang of 6 nucleotides on strand C and a 5′-phosphorylated overhang of 5 or 7 nucleotides on strand D, both complementary to a part of the overhang on strand A.
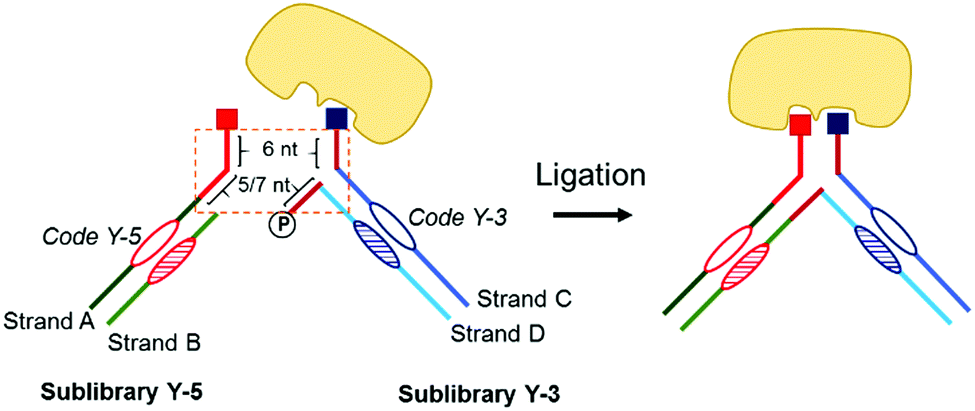 | ||
| Fig. 1 Construction of Y-EDCCL. Strands A and B are assembled to form the sub-library Y-5. Strands C and D make up the sub-library Y-3. Upon binding to the target protein, phosphorylated strand D can be ligated to strand B. | ||
The dynamic hybridization domain between Y-5 and Y-3 consists of 11 or 13 bp: 6 bp between strands A and C, and 5 or 7 bp between strands A and D. A linear 11 or 13 bp duplex would be stable at room temperature while the Y-shaped construct has a different melting behavior.23 Linear DNA melting is initiated at the extremities, and in the Y-shaped construct the DNA can also melt from the junctions, destabilizing the construct and reducing the melting temperature. The dynamics of the Y-shaped construct can be tuned by varying the length of dynamic domains. The two sub-libraries assemble in situ to form small-molecule pairs (Fig. 1). The design of the dynamic library results in fragment pairs that are constantly and randomly reshuffling in solution, but stabilized upon specific and bidentate binding to the protein target. After selection, strands B and D of the binding pairs, now in close proximity, can be ligated to allow a readout of the binding pair combination. It is important to note that the ligation step provides an additional selection mechanism, through which only the bound molecules in pairs will produce full-length DNA, thus further reducing the background noise caused by single fragment binding (Fig. 1).
In a first selection experiment, streptavidin was used as the target protein (Fig. 2). This homotetrameric protein presents four binding sites,24 making it useful for proof-of-principle experiments to test fragment-based chemical libraries.25 As the most well-known streptavidin binder, biotin has an affinity significantly stronger than any protein–drug interaction,24 therefore we instead employed biotin derivative 2-iminobiotin, with binding affinity in a range relevant for fragment-based drug discovery.26 A single iminobiotin molecule has affinity in the μM range, whereas two tethered iminobiotins show affinity in the high nM range.25
 | ||
| Fig. 2 Model selections with streptavidin and bacterial cell wall model. (a) Binding profiles of the four possible combinations from the 105-member Y-EDCCL dummy library on the streptavidin beads. The inset shows enrichment folds over the dual blank background. (b) Dual-iminobiotin enrichment from the dynamic library and static library compared to the Qmax value. The inset shows the bidentate to monodentate ratio in the dynamic library and static library. (c) Ratios between the enrichment of two iminobiotin and Qmax value of different dummy library sizes. (d) Binding profiles of the four possible combinations from the 105-member Y-EDCCL dummy library on the D-Ala–D-Ala peptide-modified solid support. | ||
Y-5 and Y-3 sub-libraries with a 5 + 6 dynamic configuration were prepared, either modified with 2-iminobiotin (Y-5-I and Y-3-I) or left unmodified (Y-5-N and Y-3-N) (Fig S9 and S12a, ESI†). A 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-member dummy library was prepared via mixing the Y-5 sub-library (1 equivalent of Y-5-I and 320 equivalents of Y-5-N) with the Y-3 sub-library (1 equivalent of Y-3-I and 320 equivalents of Y-3-N). Y-5-I, Y-5-N, Y-3-I, and Y-3-N all carry distinct codes. The fragments and the ligated products can be amplified using different PCR primers (Fig. S15, ESI†). In this experiment, 1 eq. corresponds to 200 fmol in 200 μL (1 nM). As a control, the 100000-member library was also prepared in a static, non-reshuffling format (ESI† 4.1). The experimental workflow consisted of incubating streptavidin beads with the library, washing off non-binding members, enzymatically ligating strands B and D, eluting the binders, and then analysing the product via the quantitative polymerase chain reaction (qPCR) (Fig. S13, ESI†). The amounts of each DNA could be quantified by comparing qPCR data with the calibrated DNA concentration curves (Fig. S16, ESI†).27 As expected, the dual-iminobiotin pair was the most enriched (Fig. 2a). It is more interesting, however, to view these results in comparison to selection with the non-dynamic constructs (Fig. 2b). Considering a random pairing process, it is simple to calculate the theoretical maximum amount (Qmax) of a fragment pair selected from a static library. Thus, the utility of a dynamic library can be evaluated by comparing either with a static library control or with the Qmax value. In this case, 2.82 fmol of dual iminobiotin was selected from the dynamic library versus 0.235 fmol from the static library, a 12-fold increased enrichment. The dynamic library enrichment was 4.3 times greater than the Qmax value (0.625 fmol). This result demonstrates that Y-EDCCL can increase the yield of potent binders in selection. The higher selection rate of dual-iminobiotin vs. single-iminobiotin constructs is noteworthy when the dynamic library is used, demonstrating the system's ability to convert weaker mono-valent binders to stronger bidentate binders (Fig. 2c). For a static ESAC library, although the mono-dentate binding is weaker than the bidentate binding, the binders in the “wrong” pairing are in large excess compared to the “correct” one. Y-EDCCL leads to a high signal-to-noise ratio through (1) shifting the binders from the mono-dentate to bidentate form, and (2) only the bidentate form will be ligated and analysed.
000-member dummy library was prepared via mixing the Y-5 sub-library (1 equivalent of Y-5-I and 320 equivalents of Y-5-N) with the Y-3 sub-library (1 equivalent of Y-3-I and 320 equivalents of Y-3-N). Y-5-I, Y-5-N, Y-3-I, and Y-3-N all carry distinct codes. The fragments and the ligated products can be amplified using different PCR primers (Fig. S15, ESI†). In this experiment, 1 eq. corresponds to 200 fmol in 200 μL (1 nM). As a control, the 100000-member library was also prepared in a static, non-reshuffling format (ESI† 4.1). The experimental workflow consisted of incubating streptavidin beads with the library, washing off non-binding members, enzymatically ligating strands B and D, eluting the binders, and then analysing the product via the quantitative polymerase chain reaction (qPCR) (Fig. S13, ESI†). The amounts of each DNA could be quantified by comparing qPCR data with the calibrated DNA concentration curves (Fig. S16, ESI†).27 As expected, the dual-iminobiotin pair was the most enriched (Fig. 2a). It is more interesting, however, to view these results in comparison to selection with the non-dynamic constructs (Fig. 2b). Considering a random pairing process, it is simple to calculate the theoretical maximum amount (Qmax) of a fragment pair selected from a static library. Thus, the utility of a dynamic library can be evaluated by comparing either with a static library control or with the Qmax value. In this case, 2.82 fmol of dual iminobiotin was selected from the dynamic library versus 0.235 fmol from the static library, a 12-fold increased enrichment. The dynamic library enrichment was 4.3 times greater than the Qmax value (0.625 fmol). This result demonstrates that Y-EDCCL can increase the yield of potent binders in selection. The higher selection rate of dual-iminobiotin vs. single-iminobiotin constructs is noteworthy when the dynamic library is used, demonstrating the system's ability to convert weaker mono-valent binders to stronger bidentate binders (Fig. 2c). For a static ESAC library, although the mono-dentate binding is weaker than the bidentate binding, the binders in the “wrong” pairing are in large excess compared to the “correct” one. Y-EDCCL leads to a high signal-to-noise ratio through (1) shifting the binders from the mono-dentate to bidentate form, and (2) only the bidentate form will be ligated and analysed.
In order to test the technology's performance with larger library sizes, libraries were generated using 1000- and 10000-fold excesses of Y-3-N and Y-5-N, resulting in dummy libraries of 1 million and 100 million members, respectively. The amount of iminobiotin-DNA in these libraries was 10 times reduced, with 1 equivalent corresponding to 20 fmol. Remarkably, the enrichments of the dual iminobiotin construct using 1 million and 100 million member libraries were 14.5 and 26 times greater than their corresponding Qmax values, respectively (Fig. 2c). Additionally, we prepared a small library of 1000 compounds (1 equivalent corresponding to 2 pmol). A low enrichment/Qmax ratio of 2.47 was obtained.
To demonstrate Y-EDCCL's application across target classes, a second model was employed (Fig. 2d). In this case, the target was the D-Ala–D-Ala motif from the cell wall construction process in Gram-positive bacteria, which is targeted by the antibiotic vancomycin (Fig. S12b, ESI†).28 The vancomycin and D-Ala–D-Ala interaction has been used as a model of multi-valent binding.29 Vancomycin was attached to both Y-5 and Y-3 constructs (Y-5-V and Y-3-V), and then mixed with 320 equivalents of unmodified constructs (Y-5-N and Y-3-N) to form a 103041-member library. Ac-Lys–D-Ala–D-Ala was coupled to NHS-functionalized beads via the amino group at the ε position of Lys. Dynamic selection was carried out as before. The vancomycin pair was strongly enriched (5.90 fmol), as compared to 0.032 fmol and 0.273 fmol for the two forms with single vancomycin, and 0.223 fmol for the dual-blank construct (Fig. 2d). This reflects 5.9 × 104-fold, 6.9 × 103-fold and 2.7 × 106-fold higher enrichment factor for the bivalent form, compared to the two mono-valent forms and the dual-blank construct, respectively. Moreover, the actual amount of the dual-vancomycin construct found in the dynamic selection was 9.4 times greater than the Qmax, which reflects the maximal amount from a selection using a static library (Fig. 2d). Interestingly, similar to the streptavidin–iminobiotin system (Fig. 2a), the enrichment of the mono-valent binder is higher when vancomycin is conjugated to Y-3. The highly enriched bidentate forms can be efficiently ligated, demonstrating that neither the chemical modifications nor the binding to targets on the solid support affect the ligation step. Thus, the difference between Y-5 and Y-3 indicates that the two modifications are not fully symmetric in binding. DNA is not chemically inert and can cause non-specific interactions, while its structure can also affect the ligand orientation. Dumelin and co-workers have previously shown that the linker structure between DNA and a small molecule could provide valuable information for the design of a small molecular binder.30 For DNA strands below the persistence length, the local conformation could have remarkable effect on the circularization efficacy.31,32 Although these are artifacts caused by DNA, how to translate the information associated with a particular DECL configuration to the design of small-molecule binders will be of great interest for future research.
To test Y-EDCCL in a form of affinity maturation library involving two different chemical moieties in bidentate binding, we prepared Y-EDCCL with 285 members of the Y-3 sub-library and CBS (4-carboxybenzene sulfonamide), a known binder to carbonic anhydrase II (CA II),33 on Y-5 (Fig. S12c, ESI†). The construction of the Y-3 sub-library and Y5 from the single-stranded DECL library is described in ESI† 4.2. To optimize the dynamic configuration, we compared the 6 + 5 and 6 + 7 configurations using the iminobiotin–streptavidin system. The 6 + 7 configuration showed a higher enrichment factor than the 6 + 5 configuration (Fig. S10, ESI†).
Therefore, we have chosen the 6 + 7 configuration for the library design. The Y-3 sub-library (285 nM, 1 nM for each member) and 1 nM CBS-Y-5 were mixed and the selection was performed on CA II resin (Fig. 3a). It results in a competitive condition among the Y-3 library members to form a stable complex with CBS-Y-5 to bind to CA II, reducing the binding of CBS-Y-5 in the “wrong” pairing. After ligation and PCR amplification of the eluted product, the amplicon was subjected to next generation sequencing and the result is shown in Fig. 3a. Five compounds were selected and conjugated to a DNA strand, and the binding affinity of CA II to CBS-DNA in combination with one of the five ligands was tested on an interferometry biosensor (Fig. 3b).34 The compound 99 presents a false positive, and the other 4 compounds show improvement of binding to CA II, as compared to the mono-valent binding of CA II to CBS-DNA. In particular, the combination of CBS with compound 141 increased the Kd value of CBS from 1.02 ± 0.013 μM to 140 ± 2.56 nM.
 | ||
| Fig. 3 Carbonic anhydrase II affinity maturation selection using CBS as a known binder (Y-5) and a library of 285 compounds (Y-3). (a) Evaluation of the selection against CA II by NGS. (b) Kd values of different fragments paired with CBS to CA II by a interferometry biosensor. (c–e) CA II inhibition assay using the CBS-linker derivatives (c), the CBS-linker-60 derivatives (d) and the CBS-linker-157 derivatives (e). | ||
To demonstrate that the fragments could be linked to improve the inhibitory potency, compounds 60 and 157 were conjugated to CBS using three different linkers (Fig. 3c–e). The carbonic anhydrase inhibition assay35 (ESI† 7) showed that most conjugates have higher inhibitory potency when compared with CBS and, in particular, an improved inhibition of more than 10 folds was measured for the conjugates CBS-L2-157 and CBS-L3-157 (Fig. 3e). Conjugating only the linker to CBS did not improve the inhibition remarkably (Fig. 3c).
Our immune system continuously generates antigen receptors with an almost infinite range of specificities by using the elegant DNA recombination mechanism to assemble and join gene segments irreversibly. As a fragment-based drug discovery approach, the self-assembled DNA-encoded chemical library technology aims to mimic this process to identify small-molecular binders to protein targets. DNA duplexes are used to display combinations of two chemical moieties, and the base pairing can be adjusted to permit dynamic reshuffling until stabilized by bidentate binding to the target protein. Here we have presented a novel DNA-encoded chemical library architecture, which aims to mimic the mechanisms of immunity to evolve binders through recombination, dynamics and adaption. The ligation step mimics the gene segment joining process and is chemically equivalent to the equilibrium-freezing step in dynamic combinatorial chemistry, which is often realized through photo-crosslinking, changing pH36 or reductive amination.37 Recently, photo-crosslinking has been successfully used in dynamic DECL.38 The dynamic nature and code-joining process showed significant improvement in enrichment of potent bidentate binders compared to static counterparts for libraries of multiple sizes and against different targets. On the other hand, while normal DECLs often show increased noise with the increasing library size,21 the dynamic construct exhibits a stronger relative signal for bidentate binders with the increasing size. Taken together, these data demonstrate that Y-EDCCL is a powerful tool for dynamic, dual pharmacophore DECL selection in fragment-based drug discovery. Future research will focus on optimizing the dynamic architecture, as well as carrying out selection experiments with more pharmacologically relevant protein targets.
Conflicts of interest
DyNAbind GmbH is commercializing the technologies and libraries reported in this publication. F. V. Reddavide, Y. Zhang and M. Thompson are shareholders of DyNAbind GmbH.Notes and references
- A. M. Davis, A. T. Plowright and E. Valeur, Nat. Rev. Drug Discovery, 2017, 16, 681–698 CrossRef CAS PubMed.
- D. G. Brown and J. Bostrom, J. Med. Chem., 2018, 61, 9442–9468 CrossRef CAS PubMed.
- M. Leveridge, C. W. Chung, J. W. Gross, C. B. Phelps and D. Green, SLAS discovery: advancing life sciences R & D, 2018 Search PubMed.
- G. KÖHler and C. Milstein, Nature, 1975, 256, 495 CrossRef.
- G. P. Smith, Science, 1985, 228, 1315–1317 CrossRef CAS PubMed.
- Z. Elgundi, M. Reslan, E. Cruz, V. Sifniotis and V. Kayser, Adv. Drug Delivery Rev., 2017, 122, 2–19 CrossRef CAS PubMed.
- Z. Gong, G. Hu, Q. Li, Z. Liu, F. Wang, X. Zhang, J. Xiong, P. Li, Y. Xu, R. Ma, S. Chen and J. Li, Curr. Drug Discovery Technol., 2017, 14, 216–228 CAS.
- D. Neri and R. A. Lerner, Annu. Rev. Biochem., 2018, 87, 479–502 CrossRef CAS PubMed.
- S. Brenner and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5381–5383 CrossRef CAS.
- C. Zambaldo, S. Barluenga and N. Winssinger, Curr. Opin. Chem. Biol., 2015, 26, 8–15 CrossRef CAS PubMed.
- R. A. Goodnow, Jr., C. E. Dumelin and A. D. Keefe, Nat. Rev. Drug Discovery, 2017, 16, 131–147 CrossRef PubMed.
- S. Barluenga, C. Zambaldo, H. A. Ioannidou, M. Ciobanu, P. Morieux, J. P. Daguer and N. Winssinger, Bioorg. Med. Chem. Lett., 2016, 26, 1080–1085 CrossRef CAS PubMed.
- S. Melkko, J. Scheuermann, C. E. Dumelin and D. Neri, Nat. Biotechnol., 2004, 22, 568–574 CrossRef CAS PubMed.
- M. Marczynke, K. Groger and O. Seitz, Bioconjugate Chem., 2017, 28, 2384–2392 CrossRef CAS PubMed.
- F. V. Reddavide, W. Lin, S. Lehnert and Y. Zhang, Angew. Chem., Int. Ed. Engl., 2015, 54, 7924–7928 CrossRef CAS PubMed.
- G. Li, W. Zheng, Z. Chen, Y. Zhou, Y. Liu, J. Yang, Y. Huang and X. Li, Chem. Sci., 2015, 6, 7097–7104 RSC.
- D. R. Halpin and P. B. Harbury, PLoS Biol., 2004, 2, 1022–1030 CAS.
- M. Wichert, N. Krall, W. Decurtins, R. M. Franzini, F. Pretto, P. Schneider, D. Neri and J. Scheuermann, Nat. Chem., 2015, 7, 241–249 CrossRef CAS PubMed.
- S. Melkko, Y. Zhang, C. E. Dumelin, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2007, 46, 4671–4674 CrossRef CAS PubMed.
- P. Frei, R. Hevey and B. Ernst, Chemistry, 2019, 25, 60–73 CAS.
- A. L. Satz, R. Hochstrasser and A. C. Petersen, ACS Comb. Sci., 2017, 19, 234–238 CrossRef CAS PubMed.
- A. L. Satz, ACS Comb. Sci., 2016, 18, 415–424 CrossRef CAS PubMed.
- J. B. Lee, A. S. Shai, M. J. Campolongo, N. Park and D. Luo, ChemPhysChem, 2010, 11, 2081–2084 CrossRef CAS PubMed.
- L. Chaiet and F. J. Wolf, Arch. Biochem. Biophys., 1964, 106, 1–5 CrossRef CAS PubMed.
- S. Melkko, C. E. Dumelin, J. Scheuermann and D. Neri, Chem. Biol., 2006, 13, 225–231 CrossRef CAS PubMed.
- N. M. Green, Biochem. J., 1966, 101, 774–780 CrossRef CAS PubMed.
- Y. Li, G. Zimmermann, J. Scheuermann and D. Neri, ChemBioChem, 2017, 18, 848–852 CrossRef CAS PubMed.
- H. R. Perkins, Biochem. J., 1969, 111, 195–205 CrossRef CAS PubMed.
- J. Rao, J. Lahiri, L. Isaacs, R. M. Weis and G. M. Whitesides, Science, 1998, 280, 708–711 CrossRef CAS PubMed.
- C. E. Dumelin, S. Trussel, F. Buller, E. Trachsel, F. Bootz, Y. Zhang, L. Mannocci, S. C. Beck, M. Drumea-Mirancea, M. W. Seeliger, C. Baltes, T. Muggler, F. Kranz, M. Rudin, S. Melkko, J. Scheuermann and D. Neri, Angew. Chem., Int. Ed., 2008, 47, 3196–3201 CrossRef CAS PubMed.
- B. Joffroy, Y. O. Uca, D. Presern, J. P. K. Doye and T. L. Schmidt, Nucleic Acids Res., 2018, 46, 538–545 CrossRef CAS PubMed.
- R. Vafabakhsh and T. Ha, Science, 2012, 337, 1097–1101 CrossRef CAS PubMed.
- A. Carotti, C. Raguseo, F. Campagna, R. Langridge and T. E. Klein, Quant. Struct.-Act. Relat., 1989, 8, 1–10 CrossRef CAS.
- W. Lin, F. V. Reddavide, V. Uzunova, F. N. Gur and Y. Zhang, Anal. Chem., 2015, 87, 864–868 CrossRef CAS PubMed.
- Y. Pocker and J. T. Stone, Biochemistry, 1967, 6, 668–678 CrossRef CAS PubMed.
- O. Ramstrom and J. M. Lehn, ChemBioChem, 2000, 1, 41–48 CrossRef CAS PubMed.
- I. Huc and J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2106–2110 CrossRef CAS.
- Y. Zhou, C. Li, J. Peng, L. Xie, L. Meng, Q. Li, J. Zhang, X. D. Li, X. Li, X. Huang and X. Li, J. Am. Chem. Soc., 2018, 140, 15859–15867 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc01429b |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |