
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-doped hematite thin films for electrocatalytic water oxidation in highly acidic media†
Wai Ling
Kwong
 *a,
Cheng Choo
Lee
b,
Andrey
Shchukarev
c and
Johannes
Messinger
*ac
*a,
Cheng Choo
Lee
b,
Andrey
Shchukarev
c and
Johannes
Messinger
*ac
aDepartment of Chemistry-Ångström Laboratory, Molecular Biomimetics, Uppsala University, 75120 Uppsala, Sweden. E-mail: WaiLing.Kwong@kemi.uu.se; Johannes.Messinger@kemi.uu.se
bUmeå Core Facility for Electron Microscopy, Umeå University, 90187 Umeå, Sweden
cDepartment of Chemistry, Kemiskt Biologiskt Centrum (KBC), Umeå University, 90187 Umeå, Sweden
First published on 4th April 2019
Abstract
Earth-abundant cobalt-doped hematite thin-film electrocatalysts were explored for acidic water oxidation. The strategically doped hematite produced a stable geometric current density of 10 mA cm−2 for up to 50 h at pH 0.3, as a result of Co-enhanced intrinsic catalytic activity and charge transport properties across the film matrix.
Facile conversion of renewable solar and wind energies into chemical fuel such as H2 is an enabling technology for storing and dispatching these energies on demand and for providing off-grid utilization. Burning H2 releases energy and water without carbon emission, thus H2 is in principle a ‘clean’ fuel. Central to renewable energy-driven production of H2 is the electrolysis of water into H2 and O2, where acidic proton-exchange-membrane (PEM)-based water electrolyzers are known for their advantages in compact designs and high operation efficiency, relative to their alkaline counterpart. Presently, the problem for global-scale PEM water electrolysis is the lack of low-cost (earth-abundant), stable and efficient catalysts for water oxidation, i.e., oxygen-evolution reaction (OER).
Catalysts are necessary for minimizing the kinetic barriers associated with the water splitting reactions and for suppressing undesired side reactions. Presently, the materials known for efficient and stable catalysis for OER in acidic media are limited to those based on noble metals such as Ir and Ru. However, they are extremely scarce (<0.001 ppm)1 in the Earth's crust and thus may not be able to provide a global-scale solution to our energy demand. The development of cost-effective catalysts from earth-abundant materials for OER in acidic media, however, is met with two major challenges: firstly, the OER is a kinetically demanding four-electron redox process, and therefore even the best catalysts would require a high voltammetric overpotential (>300 mV) to catalyze the OER at a rate equivalent to a current density of 10 mA cm−2 (characteristic operational current density for an ∼10% efficient solar fuel device under non-concentrated one-sun illumination);2 secondly, according to Pourbaix diagram, most earth-abundant single-metal-based materials lack innate stability against acidic corrosion at anodic potentials (viz., the applied electrical potentials necessary for OER to occur).
Various strategies have been attempted by researchers to address the above-mentioned challenges. These include the exploration of pristine as well as surface-modified transition metal-based catalysts,3–8 and the derivation of new catalysts from multi-metal oxides.9–13 The latter demonstrates the possibility of treating the catalytic activity and stability as decoupled functionalities that are facilitated by the active-yet-unstable catalytic metal and the stable-yet-inactive structural metal, respectively. Examples of such systems include CoFePbOx, NixMn1−xSb1.6−1.8Oy, MnxSb1−xOy and F-doped Cu1.5Mn1.5O4.9–11 While progress is being made toward producing earth-abundant catalysts for OER in acidic media, the catalysts reported thus far still lack competitive performances, in terms of activity and/or stability, as compared to the Ir- and Ru-based catalysts. Furthermore, the use of low-abundance (e.g., Sb) and/or toxic metals (e.g., Pb) in the catalyst composition may negatively impact the operational cost and environmental safety.
Recently, we have shown that in a mixed-polymorph catalyst film consisting of earth-abundant maghemite (γ-Fe2O3) and hematite (α-Fe2O3), the α-Fe2O3 acts as a non-catalytic scaffold that stabilizes the catalytic γ-Fe2O3 against corrosion during OER in acidic media.3 The high catalytic activity of γ-Fe2O3, as compared to α-Fe2O3, is attributed to the inherent presence of Fe-vacancy defects, which facilitates the effective charge transport across the film and the surface adsorption of reactant water. The structural fortification by α-Fe2O3 stems from its stability in acid and its lack of catalysis, which strengthen the Fe–O framework around the γ-Fe2O3 active sites. This result highlights the importance of crystal defects in facilitating the electrocatalytic activity. In our continued efforts to develop acid-stable earth-abundant OER catalysts, we introduce crystal defects in α-Fe2O3via Co doping as a strategy to activate it for OER in acidic media. Our data show that a low Co doping level significantly reduces the resistance of charge transfer for OER at the film-electrolyte interface as well as charge transport across the film. The different catalytic durability of the films of varied Co doping level suggests the important role of Co in achieving a synergistic balance in relevant factors, which include the film morphology, concentration of crystal defects and thermodynamic stability, in order to obtain a maximal catalytic durability.
In this work, monometallic iron oxide (FeOy) and bimetallic iron-cobalt oxide (CoxFe1−xOy) thin films were synthesized on Ti foil substrates by spray-pyrolysis deposition, followed by low-temperature annealing in air (see ESI† for Experimental details). The FeOy consists of interconnected, irregularly-shaped agglomerates (<1.5 μm) that are formed from individual nanograins (Fig. 1a). Smaller agglomerates are observed in the bimetallic oxide (Fig. 1b), indicating Co-induced inhibition in grain growth of the film during annealing. Aiming for minimal distortion to the crystal phase, the bimetallic oxide was synthesized at a low Co doping level (percentage of Co/(Co + Fe)), which was verified by XPS to be 5 at%, and by SEM-EDS to be 7 at% (Fig. 1c and Table S1, ESI†). For simplicity, the film is denoted as Co0.05Fe0.95Oy. The mineralogy of the films, as examined by XRD (Fig. 1d), shows only diffraction peaks assigned to α-Fe2O3.14 A shift in the XRD peak positions in the Co0.05Fe0.95Oy, relative to that in the FeOy, is hardly noticeable. However, these peaks are broader and lower in intensity, indicating a lower degree of crystallinity that may be attributed to smaller grains or crystallites, which agree with the SEM observation (Fig. 1b). Further examination of the films by Raman spectroscopy (Fig. 1e) shows the characteristic α-Fe2O3 bands,15 in agreement with the XRD results. The Raman bands at <600 cm−1 shift toward a higher wavenumber in Co0.05Fe0.95Oy, indicating a Co-induced contraction in the chemical bonds of the α-Fe2O3 structure. Also, the Raman band at 658 cm−1, which usually is ascribed to the crystal lattice disorder,16 presents in Co0.05Fe0.95Oy, corroborating the incorporation of Co into the α-Fe2O3 structure. A TEM-EDS mapping of the Co0.05Fe0.95Oy (Fig. S1a, ESI†) shows a homogeneous distribution of Fe, Co and O across the film cross-section, which complements the surface elemental mapping by SEM-EDS (Fig. S1b, ESI†). Both NBD and STEM data (Fig. 1f) show lattice spacings that correspond to the (104), (214), (220) and (012) planes of α-Fe2O3, suggesting the substitutional Co doping in the α-Fe2O3 structure.
 | ||
| Fig. 1 Top-view SEM images of (a) FeOy and (b) Co0.05Fe0.95Oy. Scale bars: 2 μm; inset 200 nm. (c) SEM-EDS of Co0.05Fe0.95Oy. (d) XRD and (e) Raman spectra of FeOy and Co0.05Fe0.95Oy. * marks the XRD peaks due to the Ti substrate. Reference XRD pattern for hematite (α-Fe2O3) also is shown. (f) STEM image and NBD pattern (inset) of Co0.05Fe0.95Oy. | ||
The catalytic performance of the films for OER in 0.5 M H2SO4 (pH 0.3) aqueous electrolyte is shown in Fig. 2. The OER activity of CoOy (characterized as amorphous Co3O4; see Fig. S2, ESI†) and IrOy also are included for comparison (see Experimental section, ESI† for synthesis details). The Co0.05Fe0.95Oy is OER-active and produces a geometric current density j = 10 mA cm−2 at an overpotential η = 650 mV, while the FeOy displays negligible j within the applied potential window of E = 1.2–2.2 V vs. RHE (Fig. 2a), which agrees with our previous study that α-Fe2O3 film is intrinsically a poor electrocatalyst for OER in acidic electrolyte.3 Although the CoOy is the most OER-active noble-metal-free catalyst in this work (inset in Fig. 2a), it exhibits only a short-term catalytic stability as indicated by the decreasing j with the increasing CV scan number, and thus is a poor catalyst for practical operation. The Co0.05Fe0.95Oy is intrinsically more active than the FeOy, as evidenced by its relatively low Tafel slope (110 mV dec−1, which approaches that of the CoOy; Fig. 2b) and a high jrsa (current density normalized by the sample real surface area; Fig. S3, ESI†). This may be explained by its enhanced surface adsorption of reactant water and an effective charge transport across the film. The latter is corroborated by the Nyquist plots shown in Fig. 2c, where two time-constants, represented by two consecutive semicircular curves, are observed (see Fig. S4, ESI† for additional Nyquist plot of FeOy). The semicircles in the low- and high-frequency region (i.e., the high- and low-Z′ region) arise due to the resistance of charge transfer at the film-electrolyte interface (Rct) and across the film (R2), respectively. Both Rct and R2 values, fitted using the equivalent circuit in Fig. S5 (ESI†), are significantly lower for the Co0.05Fe0.95Oy as compared to that of the FeOy, thus indicating that Co improves the catalytic activity and charge transport of the film, where the latter minimizes the energy loss due to potential drop across the film.
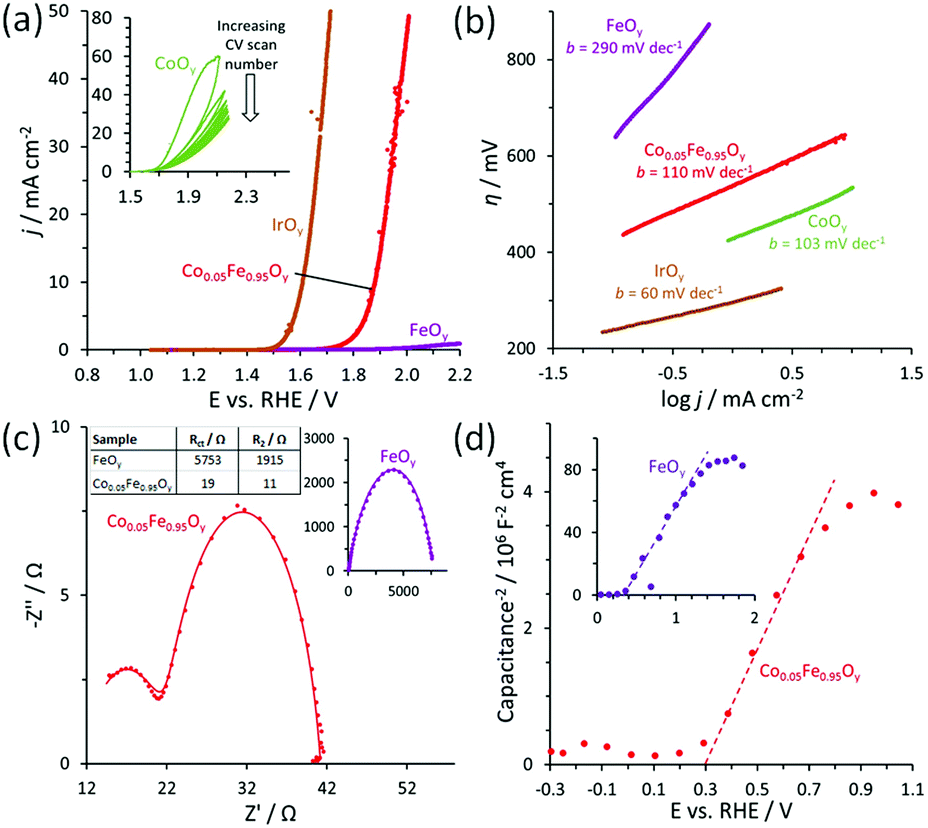 | ||
| Fig. 2 Catalytic measurements at pH 0.3. (a) Polarization curves and (b) corresponding Tafel plots (Tafel slopes b are shown). IrOy is shown as the bench-marking catalyst. (c) Nyquist plots at E = 1.89 V vs. RHE (before correction for uncompensated cell resistance). (d) Mott–Schottky plots. | ||
The surface chemical analysis by XPS (Fig. 3) shows that both FeOy and Co0.05Fe0.95Oy exhibit Fe 2p and O 1s core peaks that are similar to that of α-Fe2O3.17,18 For the Co0.05Fe0.95Oy, the peak at binding energy of 780.8 eV matches well with the Co2+ peak of CoOy (Fig. S2, ESI†) and thus suggests that the Co dopants are present as Co2+,19,20 which agrees well with assignments made in previous studies of Co-doped α-Fe2O3.21,22 The aliovalent Co doping may be charge-compensated via O vacancy formation, viz.,  , where Co×Co,
, where Co×Co, 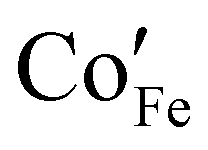 and
and 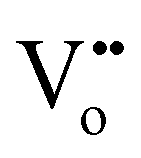 are the Co ion on the Co lattice site (neutral charge), the Co ion on the Fe lattice site (single negative charge) and the O vacancy (double positive charge), respectively. This is evidenced in the higher surface-adsorbed OH-to-lattice O peak area ratios (Table S2, ESI†) of the Co0.05Fe0.95Oy (i.e., 0.28) as compared to that of the FeOy (i.e., 0.20), where the formation of surface-adsorbed OH is attributed to the presence of O vacancies,23,24 which facilitate dissociative water adsorption.25
are the Co ion on the Co lattice site (neutral charge), the Co ion on the Fe lattice site (single negative charge) and the O vacancy (double positive charge), respectively. This is evidenced in the higher surface-adsorbed OH-to-lattice O peak area ratios (Table S2, ESI†) of the Co0.05Fe0.95Oy (i.e., 0.28) as compared to that of the FeOy (i.e., 0.20), where the formation of surface-adsorbed OH is attributed to the presence of O vacancies,23,24 which facilitate dissociative water adsorption.25
 | ||
| Fig. 3 High-resolution XPS spectra of FeOy and Co0.05Fe0.95Oy in Fe 2p, O 1s and Co 2p regions. | ||
O vacancies are known to act as donors in n-type hematite semiconductor (electron as the majority charge carrier).26 In contrast, the alternative mechanism for charge compensation via electron–hole formation (i.e., electronic compensation) would lead to a decreased donor density. To further test the charge compensation mechanism required to compensate for aliovalent doping, the donor densities of the films are probed by Mott–Schottky measurements as shown in Fig. 2d. We observe positive slopes in the linear region of the plots, meaning that both films display n-type semiconducting properties.27 The slope value, which is significantly lower upon Co-doping, is inversely proportional to the donor concentration in a semiconductor.27 This shows that the density of free electrons of the Co0.05Fe0.95Oy is remarkably higher as compared to that of the FeOy, confirming the O vacancies formation as the major charge compensation mechanism and as the factor to its improved charge transport properties. In addition, Co doping may also improve the charge transport properties by increasing the density of localized states near the Fermi level, which is supported by the theoretical work from Liao et al.28 Our observation also agrees with their calculation that Co doping improved the reaction thermodynamics of hematite for OER.28 The improvement was due to the less-positive charge of Co (2+) as compared to Fe (3+), which optimizes the binding strengths of (and thus stabilizes) the intermediate adsorbates (i.e., O*, *OH and *OOH) on the Co-doped hematite surface.
The flat-band potential Efb (obtained by extrapolating the linear curve in Fig. 2d to the x-abscissa) of the FeOy is found at 0.30 V vs. RHE while that of the Co0.05Fe0.95Oy is at 0.25 V vs. RHE. It is known that, at potentials positive of the Efb of an n-type semiconductor, a space-charge region (SCR), i.e., depletion layer, is present within the semiconductor at the semiconductor–electrolyte interface.27 The electric field associated with the SCR is crucial, particular in photoelectrocatalysis, for separating the photogenerated excitons, where the electron–holes are driven toward oxidative reaction at the semiconductor–electrolyte interface, while the electrons migrate into the semiconductor bulk. The negative shift in the Efb of the Co0.05Fe0.95Oy relative to that of the FeOy, implies that, under oxidative potential for OER (E > Efb), a stronger electric field forms in the SCR in Co0.05Fe0.95Oy, which may facilitate the flow of electrons from the surface active sites into the film matrix.
To study the effect of Co content, the bimetallic oxide films of Co doping level 2–12 at% (as determined by XPS and SEM-EDS; see Table S1, ESI†) also were synthesized. The XRD, Raman and XPS data (Fig. S6, ESI†) of the films show Co2+ substitutional doping in the α-Fe2O3 structure. A higher geometric as well as intrinsic OER activities are obtained in the bimetallic oxide film of a higher Co content (Fig. 4a and Fig. S7, ESI†). The catalytic durability, viz., overpotential, of the films to maintain j = 10 mA cm−2 is measured, as shown in Fig. 4b. While all bimetallic oxide films are OER-active, the moderately doped Co0.05Fe0.95Oy is found to be most stable (Fig. 4a), operating at pH 0.3 for up to 50 h and its overpotential rises rapidly thereafter due to corrosion, which is related to the weakening and subsequent dissolution of the metal-oxygen framework caused by proton attack. It also exhibits a near-100% Faradaic charge-to-O2 conversion efficiency (Fig. S8, ESI;† measured up to 2 h). As expected, lowering the proton concentration (viz., at pH 2) in the electrolyte results in a higher catalytic stability in Co0.05Fe0.95Oy for up to 85 h (Fig. 4b). As shown in Table S3 (ESI†), the performance of Co0.05Fe0.95Oy is comparable to that of the recently reported noble-metal-free catalysts. The relatively low catalytic durability of the more heavily doped Co0.12Fe0.88Oy (operates at pH 0.3 for up to 12 h) is possibly due to (i) its larger electrochemically active surface area (associated to its smaller agglomerate-morphology; see Fig. S9, ESI†) and thus being more exposed to proton attack, and (ii) the increased grain boundary area associated with the high number of surface defects, which are prone to corrosion.7 Since the stability of a catalyst also is influenced by the thermodynamic properties associated to the applied electrochemical potential, it is thus likely that the higher applied potential required to generate the same j by the more lightly doped Co0.02Fe0.98Oy (due to its lower OER activity) promotes transformation of the catalyst to soluble species,29,30 leading to its relatively low catalytic durability (operates at pH 0.3 for up to 28 h). Future study under in operando condition is anticipated to reveal more information regarding the structure-stability correlation in the bimetallic oxides.
 | ||
| Fig. 4 (a) Polarization curves and corresponding Tafel plots of CoxFe1−xOy. (b) Chronopotentiometric measurements of CoxFe1−xOy at j = 10 mA cm−2 and at pH 0.3 or pH 2. | ||
In summary, Co doping (2–12 at%) has been shown to be effective in activating α-Fe2O3 thin-film electrode for electrocatalytic OER via water oxidation in acidic electrolyte. Pristine α-Fe2O3 and Co3O4, on the other hand, are inactive and unstable for OER under the same operational condition, respectively. 5 at% Co-doped α-Fe2O3 shows a balanced optimizations of catalytic activity (similar level of intrinsic activity as that of Co3O4) and durability (up to 50 h and 85 h at j = 10 mA cm−2 at pH 0.3 and pH 2, respectively), while lower (2 at%) and higher (12 at%) Co doping levels result in an inferior intrinsic activity and/or durability. The catalytic activation of α-Fe2O3 is due to Co-induced improvements in the intrinsic catalytic activity and the electron transport properties across the film, which agree with the theoretical work by Liao et al. that Co mediates the binding strengths of intermediate species leading to improved OER kinetics at the Co-doped α-Fe2O3 surface.28 Furthermore, the interfacial transport of electrons from the surface active sites into the film may be enhanced by the stronger electric field associated with the depletion layer near the surface of Co-doped α-Fe2O3 as compared to pristine α-Fe2O3. The improved electron transport in the film of Co-doped α-Fe2O3 is attributed to the increased densities of free electrons as well as localized states near the Fermi level. We have shown in this work that acid-stable host materials can be catalytically activated via suitable chemical doping, and this may serve as a promising strategy to develop acid-stable OER catalysts from noble-metal-free materials.
This work was funded by Uppsala University. We thank Lars Riekehr (Uppsala University) for TEM data acquisition. We acknowledge the support from the Ångström Microstructure Laboratory and the Röntgenlab at the Department of Chemistry-Ångström at Uppsala University, and the Umeå Core Facility for Electron Microscopy (UCEM-NMI node) and XPS Platform at the Chemical Biological Centre (KBC) at Umeå University.
Conflicts of interest
There are no conflicts to declare.Notes and references
- S. R. Taylor, Geochim. Cosmochim. Acta, 1964, 28, 1273–1285 CrossRef CAS.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- W. L. Kwong, C. C. Lee, A. Shchukarev, E. Björn and J. Messinger, J. Catal., 2018, 365, 29–35 CrossRef CAS.
- J. S. Mondschein, J. F. Callejas, C. G. Read, J. Y. C. Chen, C. F. Holder, C. K. Badding and R. E. Schaak, Chem. Mater., 2017, 29, 950–957 CrossRef CAS.
- J. Wu, M. Liu, K. Chatterjee, K. P. Hackenberg, J. Shen, X. Zou, Y. Yan, J. Gu, Y. Yang, J. Lou and P. M. Ajayan, Adv. Mater. Interfaces, 2016, 3, 1500669 CrossRef.
- H. Schäfer, K. Küpper, M. Schmidt, K. Müller-Buschbaum, J. Stangl, D. Daum, M. Steinhart, C. Schulz-Kölbel, W. Han, J. Wollschläger, U. Krupp, P. Hou and X. Liu, Catal. Sci. Technol., 2018, 8, 2104–2116 RSC.
- R. Frydendal, E. A. Paoli, I. Chorkendorff, J. Rossmeisl and I. E. L. Stephens, Adv. Energy Mater., 2015, 5, 1500991 CrossRef.
- X. Yang, H. Li, A.-Y. Lu, S. Min, Z. Idriss, M. N. Hedhili, K.-W. Huang, H. Idriss and L.-J. Li, Nano Energy, 2016, 25, 42–50 CrossRef CAS.
- M. Huynh, T. Ozel, C. Liu, E. C. Lau and D. G. Nocera, Chem. Sci., 2017, 8, 4779–4794 RSC.
- I. A. Moreno-Hernandez, C. A. MacFarland, C. G. Read, K. M. Papadantonakis, B. S. Brunschwig and N. S. Lewis, Energy Environ. Sci., 2017, 10, 2103–2108 RSC.
- L. Zhou, A. Shinde, J. H. Montoya, A. Singh, S. Gul, J. Yano, Y. Ye, E. J. Crumlin, M. H. Richter, J. K. Cooper, H. S. Stein, J. A. Haber, K. A. Persson and J. M. Gregoire, ACS Catal., 2018, 8, 10938–10948 CrossRef CAS.
- P. P. Patel, M. K. Datta, O. I. Velikokhatnyi, R. Kuruba, K. Damodaran, P. Jampani, B. Gattu, P. M. Shanthi, S. S. Damle and P. N. Kumta, Sci. Rep., 2016, 6, 28367 CrossRef CAS PubMed.
- K.-L. Yan, J.-Q. Chi, J.-Y. Xie, B. Dong, Z.-Z. Liu, W.-K. Gao, J.-H. Lin, Y.-M. Chai and C.-G. Liu, Renewable Energy, 2018, 119, 54–61 CrossRef CAS.
- R. L. Blake, R. E. Hessevick, T. Zoltai and L. W. Finger, Am. Mineral., 1966, 51, 123–129 CAS.
- N. M. A. Rashid, C. Haw, W. Chiu, N. H. Khanis, A. Rohaizad, P. Khiew and S. A. Rahman, CrystEngComm, 2016, 18, 4720–4732 RSC.
- I. V. Chernyshova, M. F. Hochella Jr and A. S. Madden, Phys. Chem. Chem. Phys., 2007, 9, 1736–1750 RSC.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The Iron Oxides. Structure, Properties, Reactions, Occurrences and Uses, Wiley-VCH, Weinheim, 2003 Search PubMed.
- S. C. Petitto and M. A. Langell, J. Vac. Sci. Technol., A, 2004, 22, 1690–1696 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- J. Wang, C. Du, Q. Peng, J. Yang, Y. Wen, B. Shan and R. Chen, Int. J. Hydrogen Energy, 2017, 42, 29140–29149 CrossRef CAS.
- Y. Hou, F. Zuo, A. Dagg and P. Feng, Angew. Chem., Int. Ed., 2013, 52, 1248–1252 CrossRef CAS PubMed.
- R. Schaub, P. Thostrup, N. Lopez, E. Lægsgaard, I. Stensgaard, J. K. Nørskov and F. Besenbacher, Phys. Rev. Lett., 2001, 87, 266104 CrossRef CAS PubMed.
- R. Ovcharenko, E. Voloshina and J. Sauer, Phys. Chem. Chem. Phys., 2016, 18, 25560–25568 RSC.
- A. Y. Al-Baitai, Doctoral, University College London, 2011 Search PubMed.
- T. J. Smart and Y. Ping, J. Phys.: Condens. Matter, 2017, 29, 394006 CrossRef PubMed.
- K. Gelderman, L. Lee and S. W. Donne, J. Chem. Educ., 2007, 84, 685–688 CrossRef CAS.
- P. Liao, J. A. Keith and E. A. Carter, J. Am. Chem. Soc., 2012, 134, 13296–13309 CrossRef CAS PubMed.
- K. A. Persson, B. Waldwick, P. Lazic and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 235438 CrossRef.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc01369e |
| This journal is © The Royal Society of Chemistry 2019 |