
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile preparation of functional cycloalkynes via an azide-to-cycloalkyne switching approach†
Suguru
Yoshida
 *a,
Tomoko
Kuribara
a,
Harumi
Ito
ab,
Tomohiro
Meguro
a,
Yoshitake
Nishiyama
a,
Fumika
Karaki‡
a,
Yasutomo
Hatakeyama
a,
Yuka
Koike
c,
Isao
Kii
bc and
Takamitsu
Hosoya
*ad
*a,
Tomoko
Kuribara
a,
Harumi
Ito
ab,
Tomohiro
Meguro
a,
Yoshitake
Nishiyama
a,
Fumika
Karaki‡
a,
Yasutomo
Hatakeyama
a,
Yuka
Koike
c,
Isao
Kii
bc and
Takamitsu
Hosoya
*ad
aLaboratory of Chemical Bioscience, Institute of Biomaterials and Bioengineering, Tokyo Medical and Dental University (TMDU), 2-3-10 Kanda-Surugadai, Chiyoda-ku, Tokyo 101-0062, Japan. E-mail: s-yoshida.cb@tmd.ac.jp; thosoya.cb@tmd.ac.jp
bLaboratory for Pathophysiological and Health Science, RIKEN Center for Biosystems Dynamics Research (BDR), 6-7-3 Minatojima-minamimachi, Chuo-ku, Kobe 650-0047, Japan
cCommon Facilities Unit, Compass to Healthy Life Research Complex Program, RIKEN Cluster for Science, Technology and Innovation Hub, 6-7-3 Minatojima-minamimachi, Chuo-ku, Kobe 650-0047, Japan
dLaboratory for Chemical Biology, RIKEN Center for Biosystems Dynamics Research (BDR), 6-7-3 Minatojima-minamimachi, Chuo-ku, Kobe 650-0047, Japan
First published on 28th February 2019
Abstract
A facile method for preparing various functional cycloalkynes, including proteins incorporated with a cycloalkyne moiety, from the corresponding azides is developed. Treatment of diynes bearing strained and terminal alkyne moieties with a copper salt enabled terminal alkyne-selective click conjugation with azides, whereas a more azidophilic strained alkyne moiety was transiently protected from the click reaction via complexation with copper.
Copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC)1 and strain-promoted azide–alkyne cycloaddition (SPAAC)2,3 are trailblazing reactions of click chemistry4–6 widely used to reliably conjugate molecules in various disciplines, including materials chemistry,7 pharmaceutical sciences,8 and chemical biology.9 In particular, SPAAC is one of the most convenient methods widely applied to functionalize a broad range of molecules. This popularity stems from its applicability even in the presence of complex mixtures of molecules such as biological samples. However, preparation of cycloalkynes bearing a functional moiety is often troublesome because of the high reactivity of strained alkyne moieties and the limited number of available conjugation methods that depend on the functional moiety.
To render various cycloalkynes more easily synthesizable, we previously developed a transient protection method for cyclooctynes (Fig. 1).5h Normally, the reaction of dibenzo-fused cyclooctyne 1a bearing a terminal alkyne moiety with azide 2a proceeds exclusively towards the more azidophilic strained alkyne moiety to afford SPAAC product 3 (Fig. 1A). We found that pre-treatment of diyne 1a with a cationic copper salt resulted in the formation of a cyclooctyne–copper complex, which enabled selective CuAAC conjugation at the terminal alkyne moiety. Removal of the copper salt from the complex using aqueous ammonia afforded cyclooctyne 4a, which is the product click-conjugated at the less azidophilic terminal alkyne moiety of diyne 1a (Fig. 1B). Herein, we demonstrate that further optimization of this protection method enables a facile preparation of a broad range of functional cycloalkynes, including proteins incorporated with a cycloalkyne moiety, from the corresponding functional azides.
 | ||
| Fig. 1 Selective click reactions of diynes bearing strained and terminal alkyne moieties. TBTA = tris((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)amine and THPTA = tris((1-(3-hydroxypropyl)-1H-1,2,3-triazol-4-yl)methyl)amine. FG = functional group. | ||
We screened for conditions that efficiently afforded click-conjugated bicyclo[6.1.0]non-4-yne (BCN)3g derivative 4b from diyne 1b and azide 2a (Table 1). The strained alkyne moiety of 1b was protected by treatment with (MeCN)4CuBF4 (2.2 equiv.) in CH2Cl2via our previously reported method.5h The subsequent addition of azide 2a triggered CuAAC at the terminal alkyne moiety because an excessive amount of copper was present in the reaction mixture. We found that using THPTA10 instead of TBTA as a ligand to accelerate CuAAC was more favorable because it could be removed easily through an aqueous workup. We next explored reagents that could deprotect copper under milder conditions compared to those using highly nucleophilic aqueous ammonia. After extensive screening of chelating reagents, we found that aqueous EDTA·2Na (80 equiv.) efficiently removed copper to afford the desired 4b in an excellent yield in a one-pot three-step manner (Table 1, entry 1). In this sequence, TBTA was also available instead of THPTA for the CuAAC step (entry 2). In contrast, when NTA·2Na or DTPA·5Na solution was used in the copper deprotection step, the yield of 3b drastically decreased (entries 3 and 4). Copper deprotection using a metal-scavenging reagent rendered the aqueous workup unnecessary. Among the examined chelating reagents immobilized onto silica-gel, SiliaMetS thiourea gave the best result (entry 5). Moreover, triphenylphosphine bound on polystyrene resin (PS–TPP) was also effective (entry 8). These aqueous workup-free methods were useful for preparing water-soluble cycloalkynes (vide infra).
|
|
||
|---|---|---|
| Entry | Chelator | Yield of 4b from 1ba (%) |
| a Yields were determined by 1H NMR analysis, unless otherwise noted. b Isolated yields. c TBTA was used instead of THPTA. EDTA·2Na = disodium ethylenediaminetetraacetate; NTA·2Na = disodium nitrilotriacetate; and DTPA·5Na = pentasodium diethylenetriaminepentaacetate. | ||
| 1 | 0.1 M aq. EDTA·2Na | Quant. (98)b |
| 2c | 0.1 M aq. EDTA·2Na | 93 |
| 3 | 0.1 M aq. NTA·2Na | 41 |
| 4 | 0.1 M aq. DTPA·5Na | Trace |
| 5 | SiliaMetS thiourea | 84b |
| 6 | SiliaMetS triamine | 29 |
| 7 | SiliaMetS imidazole | 2 |
| 8 | Resin(polystyrene)–PPh2 (PS–TPP) | 80b |

The optimized conditions were applicable to the terminal alkyne-selective click reaction of a wide range of cycloalkynes bearing a terminal alkyne moiety, as demonstrated using azide 2a (Fig. 2). For example, dibenzo-fused cyclooctyne (DBCO) 4a, which we previously prepared,5h was uneventfully prepared from diyne 1a using aqueous EDTA·2Na instead of aqueous ammonia as the deprotecting reagent. Click-modified BCN derivative 4c bearing a bis(ethyleneoxy) linker was also prepared from diyne 1c quantitatively. The clickability of the strained alkyne moiety of diyne 1d with a cyclooctyne structure less strained compared to that of BCN was also protected using the cationic copper salt, and 1,4-triazole formation followed by removal of the copper salt proceeded smoothly to afford cyclooctyne 4d in high yield. This method enabled efficient preparation of click-conjugated dibenzo-fused azacyclooctyne (DIBAC)3e4e from DIBAC derivative 1e bearing a terminal alkyne moiety when the deprotection was performed using PS–TPP.11 Furthermore, not only eight-membered cycloalkynes but also nine-membered 4,8-diazacyclononyne (DACN)3i participated in this terminal alkyne-selective click reaction to afford DACN derivative 4f from diyne 1f.
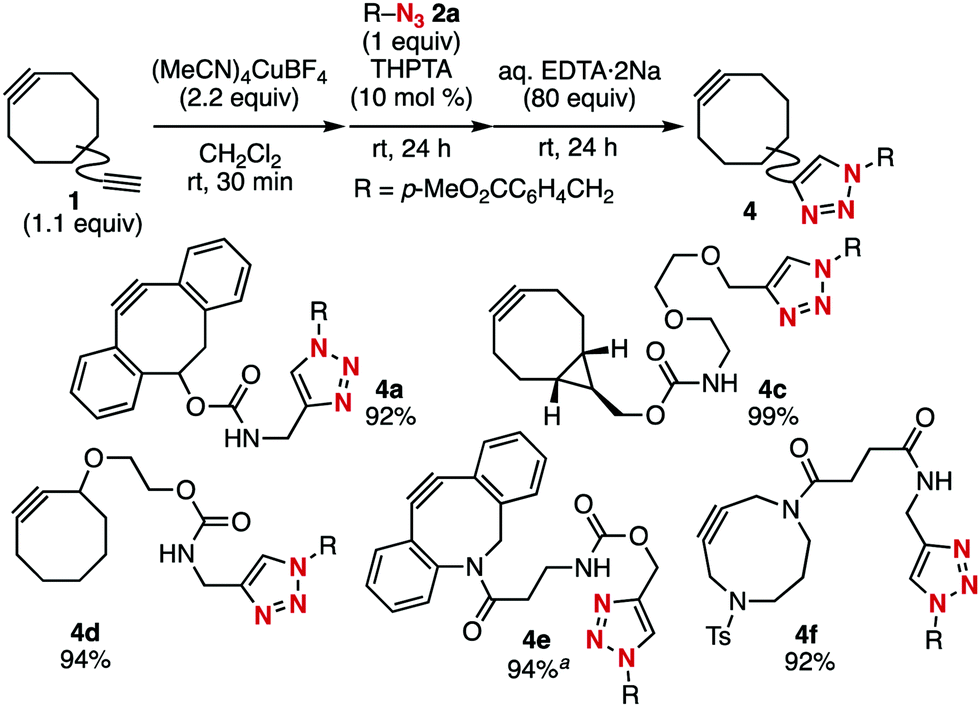 | ||
Fig. 2 Terminal alkyne-selective click conjugation of various diynes 1. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PS–TPP (160 equiv.) was used instead of aq. EDTA·2Na. PS–TPP (160 equiv.) was used instead of aq. EDTA·2Na. | ||
Using this terminal alkyne-selective click conjugation method of diynes, we prepared cycloalkynes 4g–4m bearing a diverse range of functional groups from the corresponding functional azides 2b–2h without affecting the strained alkyne and functional moieties (Fig. 3). For example, BCN–HaloTag ligand 4g was efficiently prepared from azido-HaloTag ligand 2b5c and diyne 1b. The click conjugation of biotin with DACN derivative 1f bearing a terminal alkyne moiety proceeded smoothly to afford biotin-conjugated cycloalkyne 4h in high yield. Cycloalkyne 4i, which includes a hydrophilic polyethylene moiety was also synthesized in good yield. Similarly, cycloalkynes 4j–4m conjugated with diverse fluorescent dyes such as coumarin 102, BODIPY, and tetraethylsulforhodamine were efficiently prepared from readily available fluorescent azides 2e–2h.
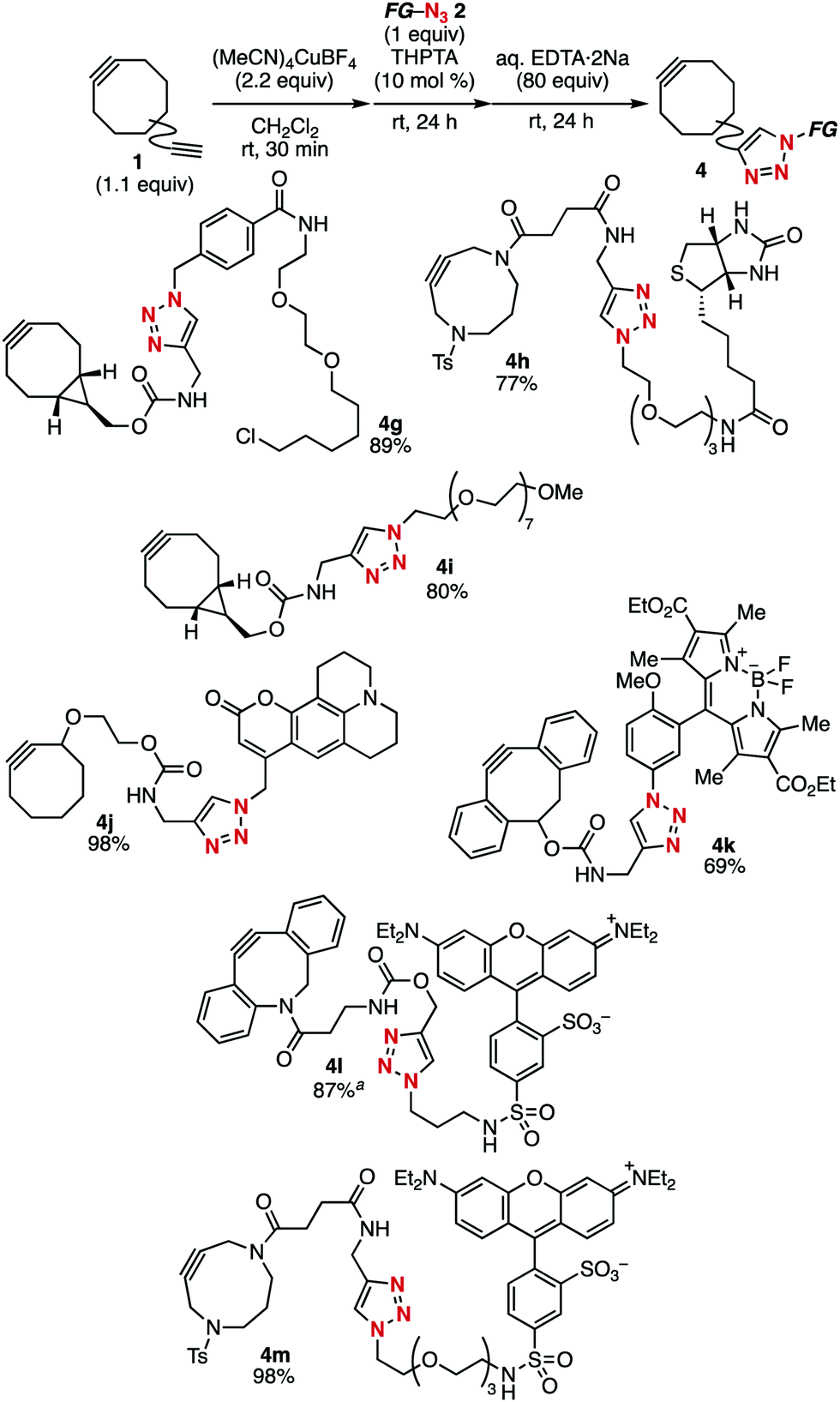 | ||
| Fig. 3 Synthesis of various functional cycloalkynes from the corresponding azides. aPS–TPP (160 equiv.) was used instead of aq. EDTA·2Na. | ||
Furthermore, Alexa Fluor 555 azide (2i), whose structure has not been disclosed by the suppliers but which is often used in biological experiments because of its favorable fluorescence characteristics, was transformed into the corresponding Alexa Fluor 555–DBCO 4nvia this method (Fig. 4A). In this case, removal of the copper salt from the reaction mixture was efficiently achieved using PS–TPP; this process did not require an aqueous workup. Treatment of HEK293 cells expressing transmembrane domain-fused HaloTag protein on the cell surface with azido-HaloTag ligand 2b followed with 4n resulted in a successful cell surface-specific fluorescent labeling by SPAAC (Fig. 4B).12 This result clearly demonstrates the utility of the azide-to-cycloalkyne switching technology.
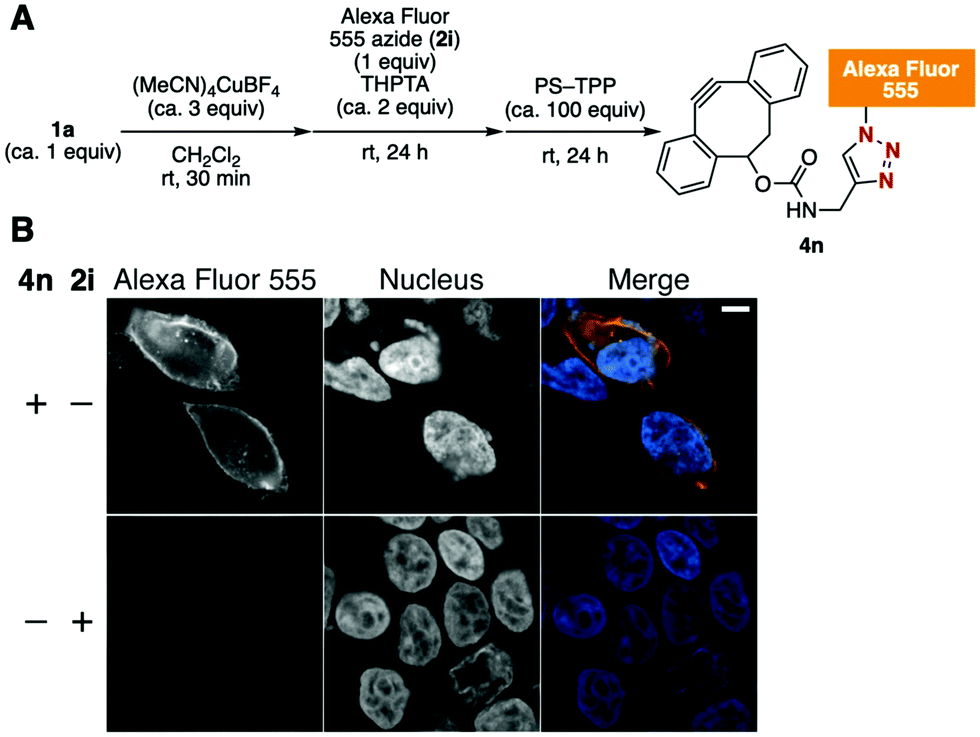 | ||
| Fig. 4 (A) Synthesis of Alexa Fluor 555–DBCO 4n. (B) Fluorescent labeling of HaloTag protein on the cell surface with 4n. HEK293 cells expressing HaloTag on the surface were incubated with azido-HaloTag ligand 2b, followed with 4n or 2i. Scale bar, 5 μm. | ||
Proteins incorporated with a cyclooctyne moiety, which were applicable to click modification with functional azides, were also easily prepared from azido-incorporated proteins, as demonstrated in the terminal alkyne-selective click conjugation using BCN-derived diyne 1c, thereby greatly expanding the utility of this method (Fig. 5A). For example, HaloTag protein-conjugated BCN was prepared from azido-HaloTag protein via the terminal alkyne-selective CuAAC reaction of copper-protected 1c under slightly modified conditions,12 followed by removal of the copper salt upon treatment with an aqueous EDTA·2Na solution. The efficiency of the transformation was determined by further modification of the HaloTag protein-conjugated BCN via the SPAAC reaction using fluorescein azide 2j. The SDS-PAGE analysis indicated that the desired fluorescein-labeled HaloTag protein was prepared in high efficiency, comparable to that prepared by the direct fluorescent labeling of HaloTag protein using fluorescein–HaloTag ligand 5 (Fig. 5B, lane 2 vs. lane 5).
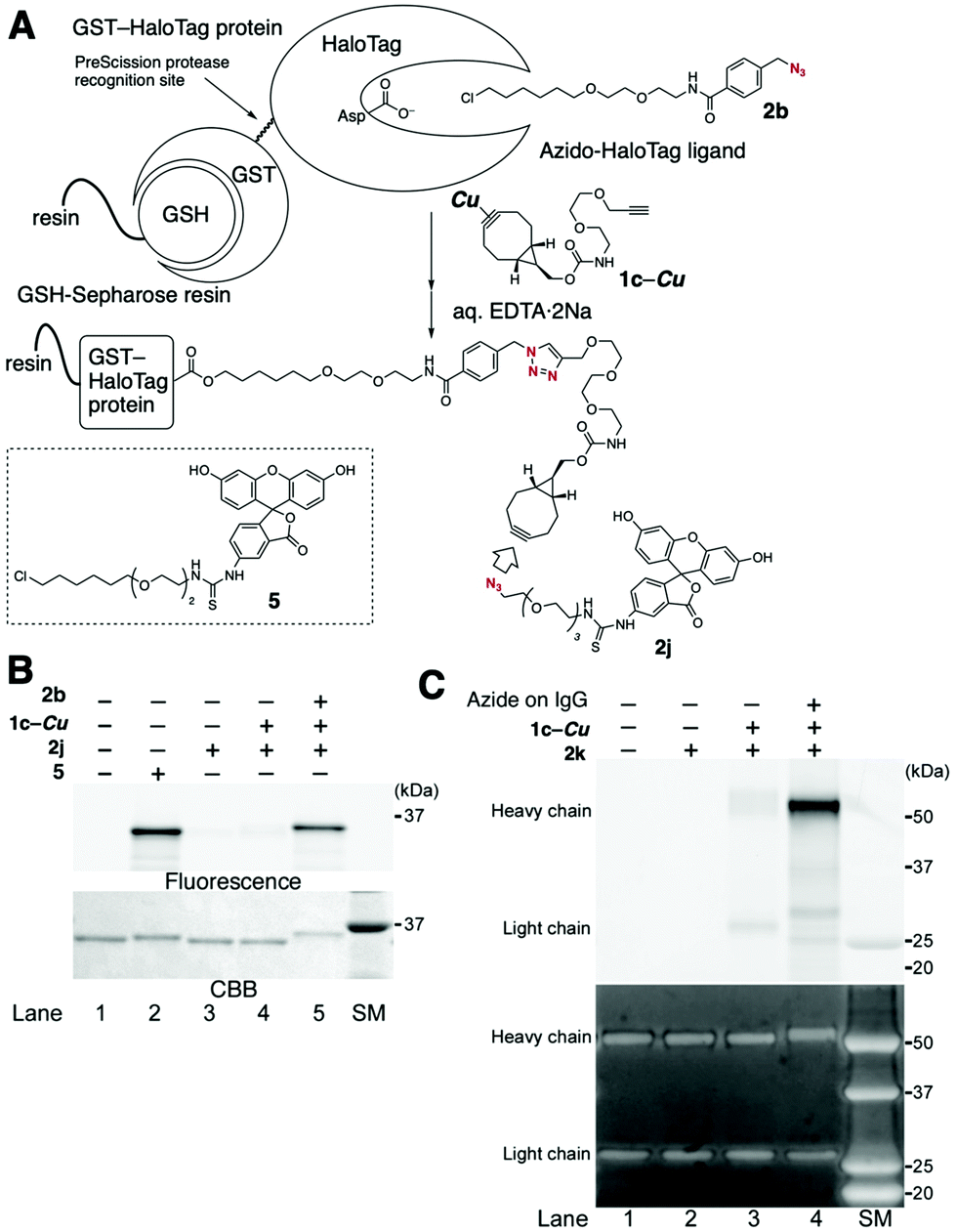 | ||
| Fig. 5 Chemical modification of azido-incorporated proteins with a fluorescent azide via BCN-conjugated proteins prepared by the terminal alkyne-selective click conjugation using Cu-protected BCN-derived diyne 1c-Cu. (A) Schematic illustration of the method. (B) Chemical modification of azido-HaloTag protein with fluorescein azide 2j. GST-HaloTag protein bound on GSH-resin was incubated in the presence of fluorescein-HaloTag ligand 5 (lane 2) or in the presence (+) or absence (−) of azido-HaloTag ligand 2b, copper-protected diyne 1c-Cu, and fluorescein azide 2j (lanes 3–5). The labeled GST-HaloTag proteins eluted from the resin were separated on SDS-PAGE. The gel was scanned using a fluorescence image analyzer and then stained with Coomassie brilliant blue (CBB). (C) Chemical modification of an azide-incorporated antibody with Alexa Fluor 488 azide (2k). An antibody installed with azido groups on its sugar chains was reacted with 1c-Cu in the presence of CuSO4, THPTA, and sodium ascorbate, followed by treatment with EDTA and Alexa Fluor 488 azide (2k). The labeled antibody was separated on SDS-PAGE, and the gel was scanned with a fluorescence image analyzer (upper panel) and then stained with One-step Ruby for the total protein detection (lower panel). SM indicates the size marker lane. | ||
Fluorescence modification of azido-incorporated cetuximab, an antibody against the human EGF receptor (EGFR), was also achieved via a similar method (Fig. 5C). In this case, an azido group was enzymatically installed onto the sugar chain at the heavy chain of IgG.12 Treatment of the azido-antibody with copper-protected 1c followed by deprotection with EDTA·2Na solution afforded antibody-conjugated BCN, which was efficiently labeled using Alexa Fluor 488 azide (2k) (Fig. 5C, lane 4). This result indicates that antibodies labeled with various functional groups are easily prepared from readily available functional azides using this system.
In summary, we have demonstrated that diverse functional cycloalkynes were easily prepared from functional azides by the terminal alkyne-selective CuAAC reaction with diynes bearing strained and alkyne moieties via transient protection of the former via complexation with copper. Chemical modification of azido-incorporated proteins with functional azides was efficiently achieved in this method. Further application studies using this method are ongoing.
This work was supported by AMED under Grant Numbers JP18am0101098 (Platform Project for Supporting Drug Discovery and Life Science Research, BINDS) and JP18am0301024 (the Basic Science and Platform Technology Program for Innovative Biological Medicine); JSPS KAKENHI Grant Numbers JP15H03118 and JP18H02104 (B; T. H.), JP16H01133 and JP18H04386 (Middle Molecular Strategy; T. H.), JP17H06414 (Organelle Zone; T. H.), JP26350971 (C; S. Y.), JP18H04568 (CMCB; I. K.), JP18J11113 (JSPS Research Fellow; T. M.), and JP17K13266 (Young Scientist B; Y. N.); the Cooperative Research Project of Research Center for Biomedical Engineering; and the Naito Foundation (S. Y.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef; (b) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (c) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed; (d) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC; (e) A. Mandoli, Molecules, 2016, 21, 1174 CrossRef PubMed.
- (a) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed; (b) M. F. Debets, C. W. J. van der Doelen, F. P. J. T. Rutjes and F. L. van Delft, ChemBioChem, 2010, 11, 1168 CrossRef CAS PubMed; (c) J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272 RSC; (d) J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed; (e) E. G. Chupakhin and M. Y. Krasavin, Chem. Heterocycl. Compd., 2018, 54, 483 CrossRef CAS.
- (a) G. Wittig and A. Krebs, Chem. Ber., 1961, 94, 3260 CrossRef CAS; (b) N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046 CrossRef CAS PubMed; (c) S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664 CrossRef CAS PubMed; (d) X. Ning, J. Guo, M. A. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253 CrossRef CAS PubMed; (e) M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97 RSC; (f) J. C. Jewett, E. M. Sletten and C. R. Bertozzi, J. Am. Chem. Soc., 2010, 132, 3688 CrossRef CAS PubMed; (g) J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422 CrossRef CAS PubMed; (h) A. Kuzmin, A. Poloukhtine, M. A. Wolfert and V. V. Popik, Bioconjugate Chem., 2010, 21, 2076 CrossRef CAS PubMed; (i) R. Ni, N. Mitsuda, T. Kashiwagi, K. Igawa and K. Tomooka, Angew. Chem., Int. Ed., 2015, 54, 1190 CrossRef CAS PubMed; (j) C. Gröst and T. Berg, Org. Biomol. Chem., 2015, 13, 3866 RSC; (k) K. Kaneda, R. Naruse and S. Yamamoto, Org. Lett., 2017, 19, 1096 CrossRef CAS PubMed; (l) E. G. Burke, B. Gold, T. T. Hoang, R. T. Raines and J. M. Schomaker, J. Am. Chem. Soc., 2017, 139, 8029 CrossRef CAS PubMed.
- (a) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (b) D. M. Patterson and J. A. Prescher, Curr. Opin. Chem. Biol., 2015, 28, 141 CrossRef CAS PubMed; (c) L.-H. Qin, W. Hu and Y.-Q. Long, Tetrahedron Lett., 2018, 59, 2214 CrossRef CAS; (d) G. Liu, J. Hu and S. Liu, Chem. – Eur. J., 2018, 24, 16484 CrossRef CAS PubMed.
- For examples for selective conjugation using azides and/or alkynes as clickable groups, see: (a) V. Aucagne and D. A. Leigh, Org. Lett., 2006, 8, 4505 CrossRef CAS PubMed; (b) P. M. E. Gramlich, S. Warncke, J. Gierlich and T. Carell, Angew. Chem., Int. Ed., 2008, 47, 3442 CrossRef CAS PubMed; (c) I. Kii, A. Shiraishi, T. Hiramatsu, T. Matsushita, H. Uekusa, S. Yoshida, M. Yamamoto, A. Kudo, M. Hagiwara and T. Hosoya, Org. Biomol. Chem., 2010, 8, 4051 RSC; (d) H. Elamari, F. Meganem, J. Herscovici and C. Girard, Tetrahedron Lett., 2011, 52, 658 CrossRef CAS; (e) S. Yoshida, A. Shiraishi, K. Kanno, T. Matsushita, K. Johmoto, H. Uekusa and T. Hosoya, Sci. Rep., 2011, 1, 82 CrossRef PubMed; (f) P. A. Ledin, F. Friscourt, J. Guo and G.-J. Boons, Chem. – Eur. J., 2011, 17, 839 CrossRef CAS PubMed; (g) D. M. Beal, V. E. Albrow, G. Burslem, L. Hitchen, C. Fernandes, C. Lapthorn, L. R. Roberts, M. D. Selby and L. H. Jones, Org. Biomol. Chem., 2012, 10, 548 RSC; (h) S. Yoshida, Y. Hatakeyama, K. Johmoto, H. Uekusa and T. Hosoya, J. Am. Chem. Soc., 2014, 136, 13590 CrossRef CAS PubMed; (i) D. N. Barsoum, N. Okashah, X. Zhang and L. Zhu, J. Org. Chem., 2015, 80, 9542 CrossRef CAS PubMed; (j) P. Gobbo, T. Romagnoli, S. M. Barbon, J. T. Price, J. Keir, J. B. Gilroy and M. S. Workentin, Chem. Commun., 2015, 51, 6647 RSC; (k) A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed; (l) R. R. Ramsubhag and G. B. Dudley, Org. Biomol. Chem., 2016, 14, 5028 RSC; (m) D. A. Sutton, S.-H. Yu, R. Steet and V. V. Popik, Chem. Commun., 2016, 52, 553 RSC; (n) M. Z. C. Hatit, J. C. Sadler, L. A. McLean, B. C. Whitehurst, C. P. Seath, L. D. Humphreys, R. J. Young, A. J. B. Watson and G. A. Burley, Org. Lett., 2016, 18, 1694 CrossRef CAS PubMed; (o) S. Yoshida, K. Kanno, I. Kii, Y. Misawa, M. Hagiwara and T. Hosoya, Chem. Commun., 2018, 54, 3705 RSC; (p) T. Meguro, S. Yoshida, K. Igawa, K. Tomooka and T. Hosoya, Org. Lett., 2018, 20, 4126 CrossRef CAS PubMed; (q) S. Yoshida, J. Tanaka, Y. Nishiyama, Y. Hazama, T. Matsushita and T. Hosoya, Chem. Commun., 2018, 54, 13499 RSC; (r) T. Yokoi, H. Tanimoto, T. Ueda, T. Morimoto and K. Kakiuchi, J. Org. Chem., 2018, 83, 12103 CrossRef CAS PubMed; (s) D. Svatunek, N. Houszka, T. A. Hamlin, F. M. Bickelhaupt and H. Mikula, Chem. – Eur. J., 2019, 25, 754 CrossRef CAS PubMed; (t) T. Yokoi, T. Ueda, H. Tanimoto, T. Morimoto and K. Kakiuchi, Chem. Commun., 2019, 55, 1891 RSC.
- For examples for selective conjugation using functional groups other than azides or alkynes, see: (a) B. C. Sanders, F. Friscourt, P. A. Ledin, N. E. Mbua, S. Arumugam, J. Guo, T. Boltje, V. V. Popik and G.-J. Boons, J. Am. Chem. Soc., 2011, 133, 949 CrossRef CAS PubMed; (b) M. R. Karver, R. Weissleder and S. A. Hilderbrand, Angew. Chem., Int. Ed., 2012, 51, 920 CrossRef CAS PubMed; (c) D. N. Kamber, L. A. Nazarova, Y. Liang, S. A. Lopez, D. M. Patterson, H.-W. Shih, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2013, 135, 13680 CrossRef CAS PubMed; (d) M. K. Narayanam, Y. Liang, K. N. Houk and J. M. Murphy, Chem. Sci., 2016, 7, 1257 RSC; (e) T. Meguro, S. Yoshida and T. Hosoya, Chem. Lett., 2017, 46, 1137 CrossRef CAS; (f) T. Meguro, N. Terashima, H. Ito, Y. Koike, I. Kii, S. Yoshida and T. Hosoya, Chem. Commun., 2018, 54, 7904 RSC.
- (a) J. Lahann, Click Chemistry for Biotechnology and Materials Science, John Wiley & Sons, West Sussex, 2009 CrossRef; (b) W. Xi, T. F. Scott, C. J. Kloxin and C. N. Bowman, Adv. Funct. Mater., 2014, 24, 2572 CrossRef CAS.
- (a) C. Wentrup, Acc. Chem. Res., 2011, 44, 393 CrossRef CAS PubMed; (b) D. Intrieri, P. Zardi, A. Caselli and E. Gallo, Chem. Commun., 2014, 50, 11440 RSC.
- (a) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905 CrossRef CAS PubMed; (b) C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075 CrossRef CAS PubMed.
- (a) T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853 CrossRef CAS PubMed; (b) X.-M. Liu, A. Thakur and D. Wang, Biomacromolecules, 2007, 8, 2653 CrossRef CAS PubMed.
- When the deprotection was performed using aq. EDTA·2Na, click conjugated DIBAC 4e was obtained in low yield (27%) due to the slow deprotection.
- See the ESI† for details.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of new compounds including NMR spectra. See DOI: 10.1039/c9cc01113g |
| ‡ Present address: Laboratory of Medicinal Chemistry, School of Pharmacy, Kitasato University, 5-9-1 Shirokane, Minato-ku, Tokyo 108-8641, Japan. |
| This journal is © The Royal Society of Chemistry 2019 |