
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sulfation made simple: a strategy for synthesising sulfated molecules†
Daniel M.
Gill
a,
Louise
Male
b and
Alan M.
Jones
 *a
*a
aSchool of Pharmacy, University of Birmingham, Edgbaston, B15 2TT, UK. E-mail: a.m.jones.2@bham.ac.uk; Tel: +44 (0)1214147288
bSchool of Chemistry, University of Birmingham, Edgbaston, B15 2TT, UK
First published on 12th March 2019
Abstract
The study of organosulfates is a burgeoning area in biology, yet there are significant challenges with their synthesis. We report the development of a tributylsulfoammonium betaine as a high yielding route to organosulfates. The optimised reaction conditions were interrogated with a diverse range of alcohols, including natural products and amino acids.
Organosulfates play a variety of important roles in biology, from xenobiotic metabolism to the downstream signalling of steroidal sulfates in disease states.1 Sulfate groups on glycosaminoglycans (GAGs) such as heparin, heparan sulfate and chondroitin sulfate, facilitate molecular interactions and protein ligand binding at the cellular surface,2 an area of interest in drug discovery.3
Heparin (an anticoagulant),4 avibactam® (a β-lactamase inhibitor),5 sotradecol (a treatment for varicose veins),6 the sulfate metabolite of paracetamol (an analgesic),7 and the glycomimetic C3 (used to study atherosclerosis),8 contain organosulfate motifs (Fig. 1). There are many other natural sources of bioactive sulfated compounds.9
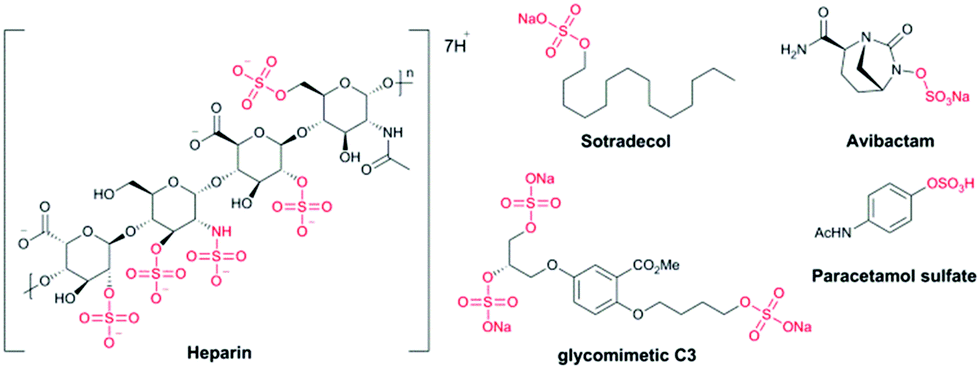 | ||
| Fig. 1 Examples of drug molecules containing organosulfates: heparin, sotradecol® and avibactam®; a chemical tool compound (C3) and a metabolite of paracetamol. | ||
The incorporation of polar hydrophilic organosulfate groups onto drug-like molecules is timely to facilitate research investigating sulfated GAGs as potential new therapies.10 However, the insertion and isolation of sulfate groups into target molecules remains a challenging aspect of their synthesis,11 prompting recent advances into sulfate revealing pro-drugs.12
The presence of one or more sulfate group makes chemical synthesis and purification of (per)sulfated compounds challenging, primarily due to their poor solubility in organic solvents.13 Therefore the insertion of organosulfate groups is typically the final step in a synthetic method, limiting further chemical modifications.14 A variety of methods to prepare organosulfates are shown in Chart 1.
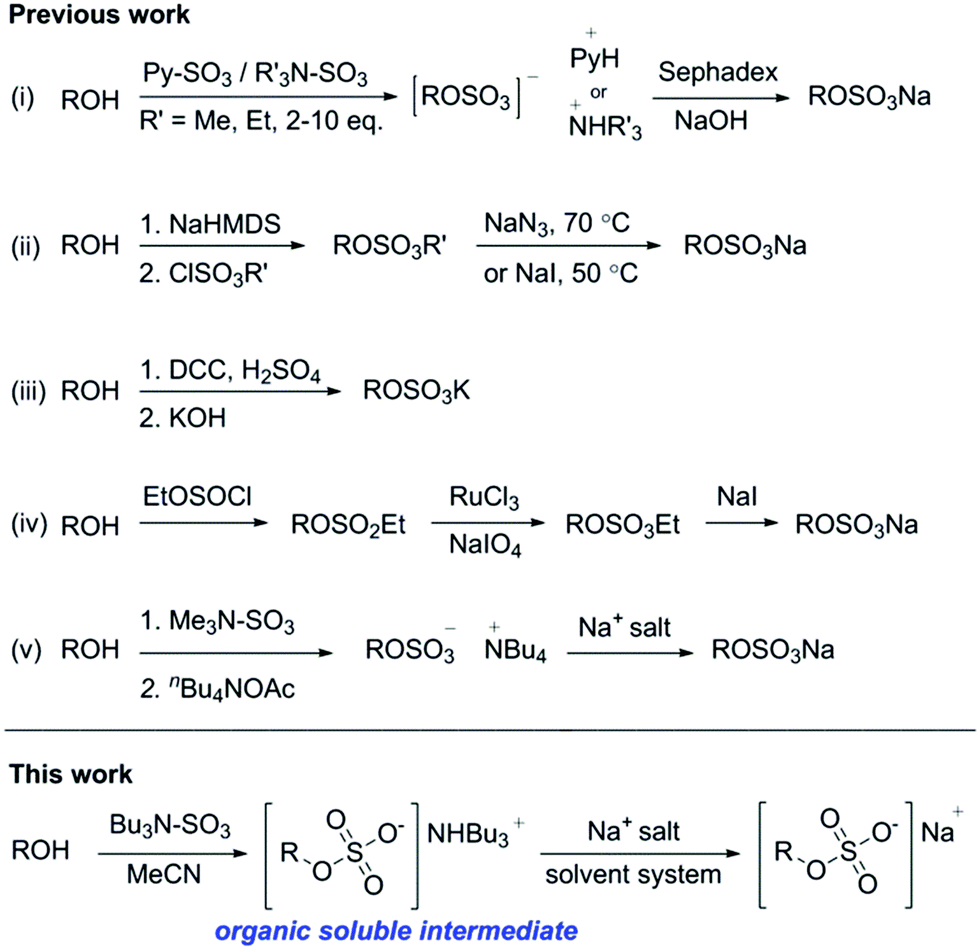 | ||
| Chart 1 Previous strategies to the synthesis of sulfate esters: this work: sulfate ester formation using tributylsulfoammonium betaine gives direct access to an organic soluble organosulfate intermediate that can be readily ion exchanged. | ||
The preparation of organosulfate esters include a microwave assisted approach to the sulfation of alcohols, using Me3NSO3 and PySO3, Chart 1(i)).15,16 The addition of catalytic diaryl borinic acid can be used with Me3NSO3 to sulfate carbohydrates.17 The limitations with these routes include the need for a stoichiometric excess of the reagent per alcohol group (up to 10 eq. per hydroxyl group) and difficulties with purification. Poor solubility in organic solvents makes aqueous purification protocols and ion-exchange chromatography a standard procedure, limiting the practicality. An alkyl chlorosulfate ester with subsequent deprotection has proven a reliable method, but limitations include the need to use a strong base and a deprotection step leads to side products (Chart 1(ii)).18 A DCC/H2SO4-sulfate coupling has been demonstrated but is not amenable to acid sensitive substrates (Chart 1(iii)).19 A sulfitylation–oxidation protocol (Chart 1(iv)) involves the synthesis of a protected sulfite ester, oxidation to the protected sulfate ester and cleavage to the sodium sulfate salt.20 However, the use of multiple steps and purification sequences is limiting. A process route to Avibactam21 (Fig. 1 and Chart 1(v) involved sulfation of the hydroxylamine intermediate using Me3NSO3, followed by cation exchange with tetrabutylammonium acetate, gave the organosulfate as its tetrabutylammonium salt. The sodium salt was obtained by precipitation in 77% yield over 2 steps on a multi-kg scale. Similarly, the use of a sulfate bis(tributylammonium) salt for the preparation for nucleoside phosphosulfates22 highlighted that the solubility of the organosulfate ester can be modulated by increasing the lipophilicity of the corresponding cation.
During a medicinal chemistry programme we encountered difficulties using the established sulfation methods to per-sulfate compounds, due to the poor solubility of the organosulfate intermediates and their resulting purification. We sought to develop an all in one reagent, to improve the solubility of the intermediate organosulfate ester. Combining tributylamine (pKa, 10.89) with SO3, we envisioned this would create a complex (Bu3NSO3, 1) for the persulfation of alcohols, with increased lipophilicity of the intermediate sulfate ester, improving the overall solubility of the organosulfates in organic solvents. We rationalised that 1 would retain similar activity to SO3 complexes with Et3N and Me3N, due to their similar Lewis base strengths, pKa = 11.01 and 10.63, respectively. Overall, 1 may permit sequential chemical steps in organic solvents, with a simple purification method to the corresponding sodium salt, streamlining the synthesis of organosulfates.
The complexation of sulfur trioxide to nitrogen or oxygen containing molecules (such as pyridine, NMe3, NEt3, DMF, THF and dioxane) is well known,11,15 the use of an organic solubilising partner, tributylamine, is however not. To the best of our knowledge, the only literature report of the synthesis and physical study of 1 was by Moede in 1949.23 It was not until 1976 that Parshikov and co-workers24 studied 1 as a sulfating agent on simple aliphatic alcohols (without spectroscopic characterisation) and found that 1 reacts via an SN2 mechanism driven by the hydrogen-bonding propensity of the alcohol under study. However, to the best of our knowledge no further use or development of this reagent has been reported.
We synthesised 1 by reaction of tributylamine with chlorosulfonic acid, affording a 90% yield on a 60 g scale (Scheme 1(a)).25 For the first time both NMR spectral data and the crystal structure of 1, obtained from small molecule single crystal X-ray diffraction, was determined (Scheme 1(b)).26 Bu3NSO3 (1) adopts a Gauche conformation within an asymmetric unit cell caused by hydrogen bonding between the methylene hydrogen atoms α to the nitrogen and the oxygens of SO3. The measured N–S bond length in 1 is 1.886(3) Å, a comparable bond length to a single N–S bond (typically: 1.73–1.83 Å versus 2.06 Å for a donor–acceptor system),27 suggesting that 1 exists as a betaine in the solid state which may have implications for the other unsolved amine–SO3 complexes and their associated mechanisms.
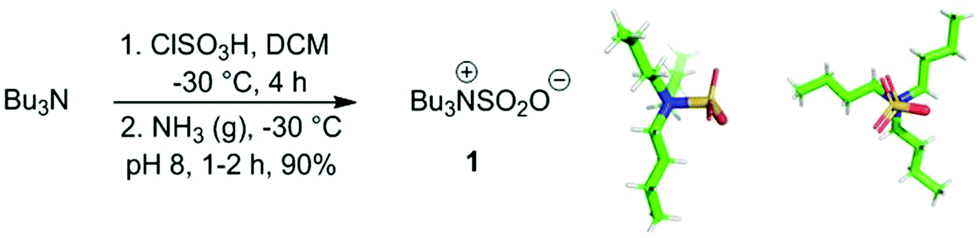 | ||
| Scheme 1 (a) Synthesis of Bu3NSO3 (1); (b) alternative views of the crystal structure of 1 obtained from small molecule single crystal X-ray diffraction. | ||
Benzyl alcohol (2a) was selected for the optimisation study (Chart 2) due to a distinct down-field shift (+0.35 ppm) of the benzylic signal after sulfation (by 1H-NMR spectroscopy).
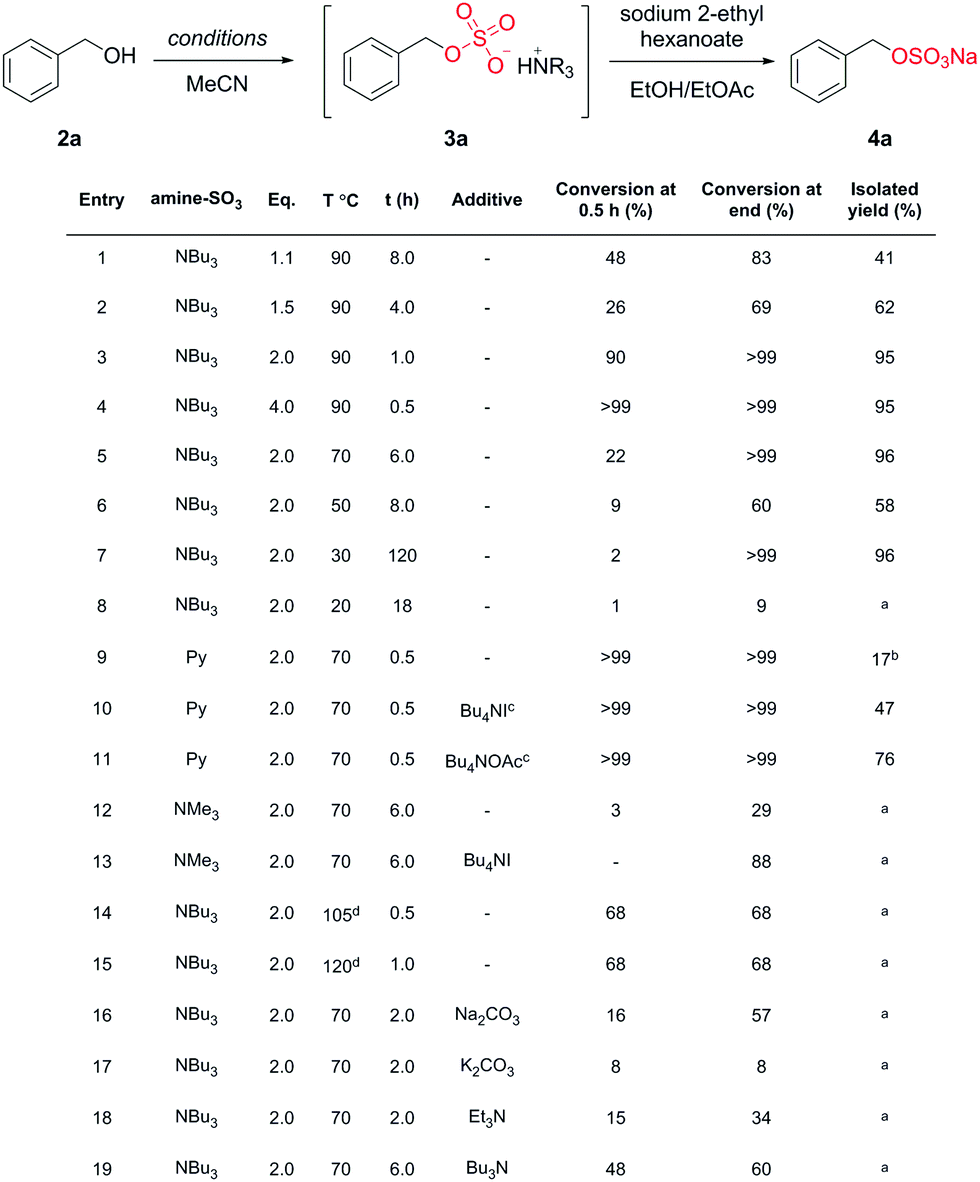 | ||
Chart 2 Optimisation of a model system. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Not isolated; bisolated as the pyridinium salt; cadditive used during work-up; dmicrowave irradiation. Not isolated; bisolated as the pyridinium salt; cadditive used during work-up; dmicrowave irradiation. | ||
We examined the sulfation of 2a with varying equivalents of 1 (entries 1 to 4). It was found that 2.0 equivalents of 1 was optimal for high conversions (>99%) and isolated yields (95%, entry 3). Less than 2.0 equivalents of 1 (entries 1 and 2) gave incomplete conversion to 3a. We next surveyed the effect of temperature (entries 5–8 vs. entry 3). 90 °C and 2.0 equivalents of 1 gave complete conversion to 3a within 2 h. Notably, reaction completion was achieved, even at 30 °C (with increased reaction time) demonstrating that 1 could be a suitable reagent for temperature sensitive substrates such as proteins.13
We compared the use of two commercially available amine–SO3 complexes, namely pyridine and trimethylamine (entries 9 and 12, respectively). Py–SO3 was more reactive than 1, giving complete conversion to 3a in 0.5 h but only a 17% isolated yield of the pyridinium species. As a control experiment, the sequential exchange of the pyridinium salt with different Bu4N cations afforded the isolation of 4a in 47% (Bu4NI) and 76% (Bu4NOAc) yield, respectively (entries 10 and 11). The use of Me3N–SO3 gave poor results (0% isolated yield of 4a from a 29% conversion to 3a). The addition of Bu4NI to the reaction mixture significantly improved the reaction, affording an 88% conversion to 3a (entry 13). Unfortunately, a more complex reaction mixture was detrimental to the isolation of 4a. These results demonstrate the isolation benefits associated with the protocol developed with 1.
We observed that 1 can achieve high conversions of 2a to 3a/4a without microwave irradiation (entries 1–7 vs. 14 and 15) unlike other reported sulfating agents. The addition of a hetero or homogenous base (entries 16–19) was investigated and in all cases this led to a decrease in conversion to 3a. Most notably, the addition of tributylamine (entry 19) initially increased the rate conversion to 3a, but reduced the overall conversion contradicting the original report.24 We rationalised that the addition of Et3N (with a higher pKa value than Bu3N) competes in the reaction, forming Et3NSO3in situ. This amine exchange has also been observed in the reaction of Et3N with PySO3.21
We applied the optimal conditions to the synthesis of a range of sulfate esters (Chart 3). In all examples (Chart 3) we observed a near-quantitative conversion to the corresponding sulfate ester as the tributylammonium salt, independent of both electronic and steric factors. Differences occurred when converting the intermediate into a crystalline sodium salt; most likely due to the variability in precipitation of the sodium salt. Therefore differences of electron-withdrawing and electron-donating factors are difficult to draw with certainty. Steric bulk adjacent to the reacting centre was accommodated with ease (4h) and the reaction could also be applied to phenolic alcohols (4i).
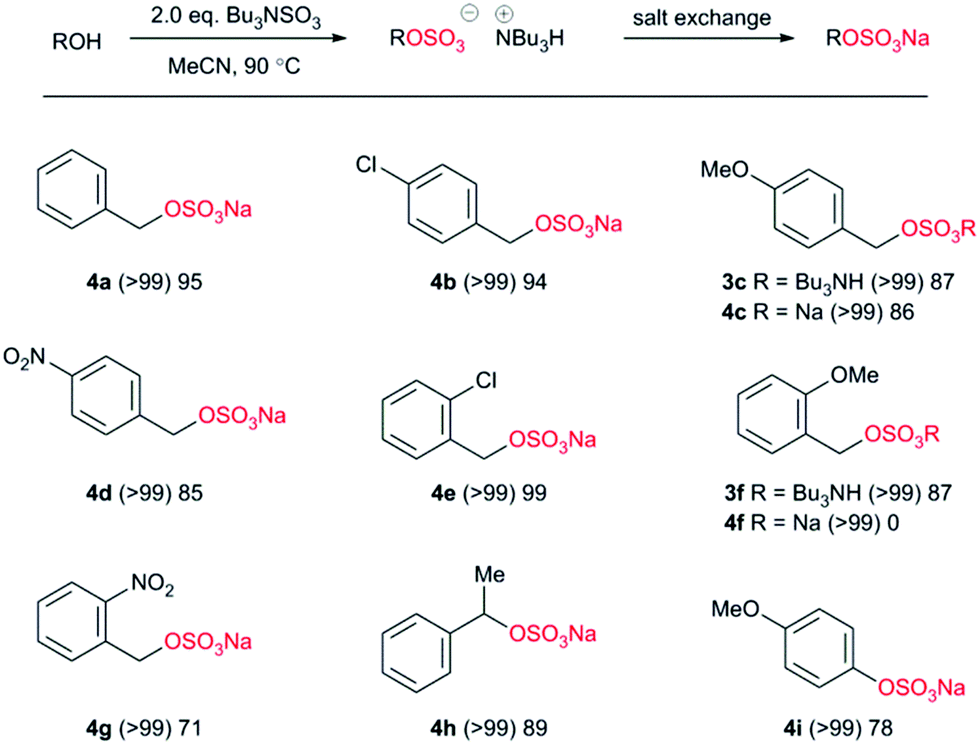 | ||
| Chart 3 Application of methodology to benzylic and phenolic alcohols. (Parentheses indicates reaction conversion as measured by 1H NMR spectroscopy.) | ||
Driving an organosulfate ester reaction to completion, by sulfating all possible hydroxyl sites in a structure remains a synthetic challenge. This is influenced by anionic crowding, making per-sulfation progressively more difficult.16 Therefore we applied the conditions to examples of compounds requiring two or more sulfation events, including natural products, and compounds containing different hydroxyl moieties within the same structure to probe the selectivity profile (Chart 4).
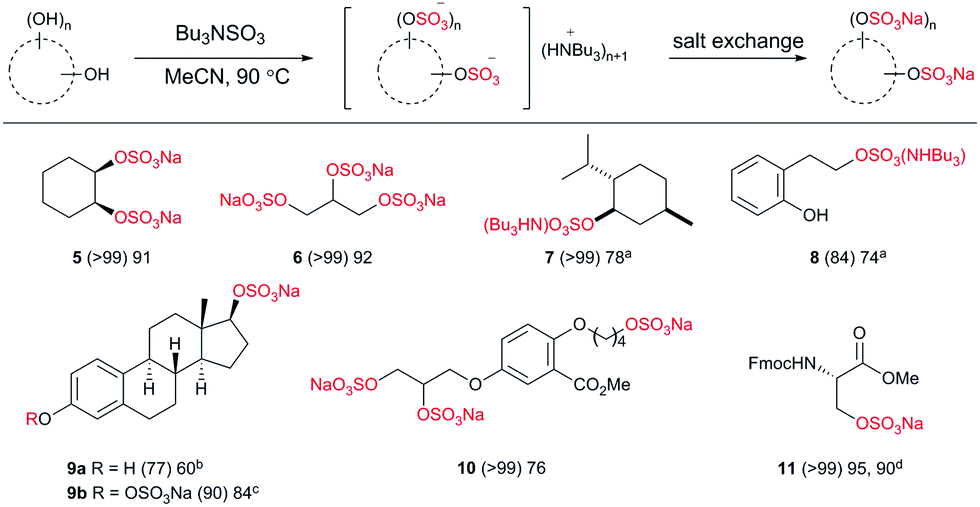 | ||
| Chart 4 Reaction scope on diverse alcohols (2.0 eq. of 1 per OH group except where indicated). aIsolated as NBu3H salt; b1.5 eq. of 1; c5.0 eq. of 1; dreaction performed at 38 °C using 4.0 eq. of 1 in DMF. (Parentheses indicate reaction conversion as measured by 1H NMR spectroscopy.) | ||
Cyclohexane-1,2-diol afforded the disulfate in high conversion and isolated yield, likewise with glycerol, the tri-sulfated analogue was prepared with ease (5 and 6, respectively). The reaction of 1 with menthol (7) delivered the NHBu3 salt with ease.
To probe the alcohol selectivity profile, a primary alcohol was functionalised in preference to a phenol in 8. Therefore, β-estradiol afforded 17-β-estradiol sulfate28 over the more common metabolite 3-β-esteradiol in 60% isolated yield (9a) using 1.5 eq. 1. Using 5.0 eq. of 1 both the 17- and 3-positions were sulfated in 84% isolated yield (9b).
We then applied the methodology to the original medicinal chemistry challenge, the sodium salt of glycomimetic C3 (10).8 High conversion and an isolated yield of 76% on a 500 mg scale was achieved. The methodology was applied to the Fmoc-protected amino acid, serine, resulting in excellent yields (95% under normal conditions) and 90% at a non-denaturing temperature (11) both using 4.0 eq. of 1. Importantly, no loss in enantiomeric ratio was observed upon sulfation (>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).29
:1).29
In summary, we have reported the first scalable preparation and reaction scoping study of 1 as a mild, bench-stable, and chromatography-free method to access organic sulfate esters as their ammonium or sodium salts. The reaction holds promise with the ability to install up to three sulfate groups on complex scaffolds including examples where sterics would limit other methods. Further work to elucidate the scope of the reaction on human sulfate metabolites is ongoing.
The authors thank Dr Chi Tsang and the Centre for Chemical and Materials Analysis in the School of Chemistry, University of Birmingham for analytical support. D. M. G. thanks the University of Birmingham for a PhD scholarship. The authors thank Dr J. W. Mueller, Dr K. A. Roper, and Ms A. M. Benedetti for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. W. Mueller, L. C. Gilligan, J. Idkowiak, W. Arlt and P. A. Foster, Endocr. Rev., 2015, 36, 526–563 CrossRef CAS PubMed; (b) P. A. Foster and J. W. Mueller, J. Mol. Endocrinol., 2018, 61, T271–T283 Search PubMed.
- D. M. Tollefsen, in Prog. Mol. Biol. Transl. Sci., ed. Zhang, L., Academic Press, 2010, vol. 93, pp 351–372 Search PubMed.
- B. Ernst and J. L. Magnani, Nat. Rev. Drug Discovery, 2009, 8, 661–677 CrossRef CAS PubMed.
- R. J. Linhardt, J. Med. Chem., 2003, 46, 2551–2564 CrossRef CAS PubMed.
- T. Stachyra, P. Levasseur, M.-C. Péchereau, A.-M. Girard, M. Claudon, C. Miossec and M. T. Black, J. Antimicrob. Chemother., 2009, 64, 326–329 CrossRef CAS PubMed.
- M. P. Goldman and R. G. Bennett, J. Am. Acad. Dermatol., 1987, 17, 167–182 CrossRef CAS PubMed.
- G. M. Pacifici and K. Allegaert, Curr. Ther. Res., 2015, 77, 24–30 CrossRef CAS PubMed.
- A. M. Mahmoud, F. L. Wilkinson, A. M. Jones, J. A. Wilkinson, M. Romero, J. Duarte and M. Y. Alexander, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 3311–3322 CrossRef CAS PubMed.
- F. Carvalhal, M. Correira-da-Silva, E. Sousa, M. Pinto and A. Kijjoa, J. Mol. Endocrinol., 2018, 61, T211–T231 Search PubMed.
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS PubMed.
- R. A. Al-Horani and U. R. Desai, Tetrahedron, 2010, 66, 2907–2918 CrossRef CAS PubMed.
- E. M. Gordon, M. A. J. Duncton and M. A. Gallop, J. Med. Chem., 2018, 61, 10340–10344 CrossRef CAS PubMed.
- S. Hemmerich, D. Verdugo and V. L. Rath, Drug Discovery Today, 2004, 9, 967–975 CrossRef CAS PubMed.
- M. Rawat, C. I. Gama, J. B. Matson and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2008, 130, 2959–2961 CrossRef CAS PubMed.
- E. E. Gilbert, Chem. Rev., 1962, 62, 549–589 CrossRef CAS.
- A. Raghuraman, M. Riaz, M. Hindle and U. R. Desai, Tetrahedron Lett., 2007, 48, 6754–6758 CrossRef CAS PubMed.
- D. Gorelik, Y. C. Lin, A. L. Briceno-Strocchia and M. S. Taylor, J. Org. Chem., 2019, 84, 900–908 CrossRef CAS PubMed.
- L. S. Simpson and T. S. Widlanski, J. Am. Chem. Soc., 2006, 128, 1605–1610 CrossRef CAS PubMed.
- R. O. Mumma, Lipids, 1966, 1, 221–223 CrossRef CAS PubMed.
- M. Huibers, Á. Manuzi, F. P. J. T. Rutjes and F. L. van Delft, J. Org. Chem., 2006, 71, 7473–7476 CrossRef CAS PubMed.
- M. Ball, A. Boyd, G. J. Ensor, M. Evans, M. Golden, S. R. Linke, D. Milne, R. Murphy, A. Telford, Y. Kalyan, G. R. Lawton, S. Racha, M. Ronsheim and S. H. Zhou, Org. Process Res. Dev., 2016, 20, 1799–1805 CrossRef CAS.
- J. Kowalska, A. Osowniak, J. Zuberek and J. Jemielity, Bioorg. Med. Chem. Lett., 2012, 22, 3661–3664 CrossRef CAS PubMed.
- J. A. Moede and C. Curran, J. Am. Chem. Soc., 1949, 71, 852–858 CrossRef CAS.
- (a) B. Passet and V. Parshikov, Org. React., 1976, 13, 555–560 CAS; (b) V. Parshikov, B. Passet and N. Kopeina, Org. React., 1976, 13, 310–314 CAS.
- The original report of 123 used the reaction of NBu3 with liquid SO3 in CCl4 but no isolated yield or characterisation data (apart from the melting point) is available.
- CCDC 1894165 (1) contains the supplementary crystallographic data for this paper. Crystal data for 1: C12H27NO3S (M = 265.40 g mol−1): trigonal, space group R3c (no. 161), a = 14.3352(2) Å, c = 12.2455(2) Å, V = 2179.28(8) Å3, Z = 6, T = 100.01(10) K, μ(CuKα) = 1.969 mm−1, Dcalc = 1.213 g cm−3, 8776 reflections measured (16.14° ≤ 2Θ ≤ 147.692°), 977 unique (Rint = 0.0273, Rsigma = 0.0115) which were used in all calculations. The final R1 was 0.0209 (I > 2σ(I)) and wR2 was 0.0570 (all data).
- (a) F. A. Kanda and A. J. King, J. Am. Chem. Soc., 1951, 73, 2315–2319 CrossRef CAS; (b) G. J. Kubas, A. C. Larson and R. R. Ryan, J. Org. Chem., 1979, 44, 3867–3871 CrossRef CAS.
- The only other report to the preparation of 9a involved a protection-deprotection cascade: H. Fex, K. E. Lundvall and A. Olsson, Acta Chem. Scand., 1968, 22, 254–264 CrossRef CAS PubMed.
- We thank reviewer 2 for this suggestion and the insight it provided. Chiral HPLC traces are provided in the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Preparative routes, compound characterisation, copies of 1H and 13C spectra and the cif file for the X-ray crystal structure of 1. CCDC 1894165 (1). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc01057b |
| This journal is © The Royal Society of Chemistry 2019 |