
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUSP7: combining tools towards selectivity†
Lorina
Gjonaj
ab,
Aysegul
Sapmaz
ab,
Román
González-Prieto
 c,
Alfred C. O.
Vertegaal
c,
Dennis
Flierman
*ab and
Huib
Ovaa
*ab
c,
Alfred C. O.
Vertegaal
c,
Dennis
Flierman
*ab and
Huib
Ovaa
*ab
aOncode Institute and Department of Cell and Chemical Biology, Leiden University Medical Center, Einthovenweg 20, 2333 ZC, Leiden, The Netherlands. E-mail: d.flierman@pepscan.com; H.Ovaa@lumc.nl
bDivision Cell Biology, The Netherlands Cancer Institute, Plesmanlaan 121, 1066 CX, Amsterdam, The Netherlands
cDepartment of Cell and Chemical Biology, Leiden University Medical Center, Einthovenweg 20, 2333 ZC, Leiden, The Netherlands
First published on 11th April 2019
Abstract
Active-site directed probes based on the ubiquitin scaffold have been successfully applied as tools to determine the levels of active deubiquitinating enzymes (DUBs). Here, we show the development of a Ub-based reagent selective for the DUB USP7. This concept can be applied for the generation of other new selective reagents.
Protein function, localization, stability and protein–protein interactions are just a few of the cellular processes regulated by ubiquitination. Not only is ubiquitination one of the most common post-translational modifications, it is arguably one of the most complex, and it is therefore tightly regulated.1 Through an elaborate enzymatic cascade consisting of three separate enzymes, ubiquitin can be attached to a specific lysine or the N-terminus of a substrate protein via its C-terminus.2 In addition, ubiquitin molecules can be linked together forming highly complex chains. Conversely, deubiquitinases (DUBs) serve to control the level of ubiquitination by catalysing the release of ubiquitin (Ub) from substrates and by taking apart different ubiquitin chains.3 To date, nearly 100 human DUBs have been identified, divided into six different families.4 USP7, a member of the ubiquitin specific protease (USP) family, has attracted great attention as a potential therapeutic target.5 Anomalous levels of this DUB have been linked to various diseases, including cancer, making USP7 an intriguing target for further study to elucidate its cellular function. Following more than a decade of drug development efforts, many selective small molecule inhibitors for USP7 have been reported.6 In addition to these small molecule inhibitors, Ub variant-based inhibitors have shown a high inhibitory effect in specific processes when overexpressed inside cells. Even though ubiquitin is a relatively small protein of only 76 amino acids, it has a fascinating conformational plasticity, allowing binding to various proteins with a wide range of functionalities and unrelated structural specificity. DUBs can recognize, and process or remove complex ubiquitin chains from ubiquitylated substrates through specific interactions between ubiquitin and various binding domains within these DUBs. These interactions can be easily removed or enhanced by overwriting the existing interactions at the interface between the two proteins; even single mutations introduced in the structure of ubiquitin can cause a large change in affinity.
One of the most established tools used to generate strong binders is phage display. This approach generates mutant Ub proteins with differing binding capabilities.7 Taking advantage of these features, libraries of ubiquitin variants proved to be efficient in targeting a number of DUBs, including USP7, as well as other Ub-specific enzymes such as E3 ligases, which are part of the previously discussed enzymatic cascade.8 The first successful example based on this technique was established by Ernst et al.7 They created libraries of phage-displayed soft-randomized Ub variants, which after rounds of selection led to a number of strong binders of a selected target protein (e.g. USP2, USP8 or USP21). In another study, authors designed libraries by combining computational studies and phage display, reporting Ub variants including U7Ub25.2540 with increased affinity for USP7.9 Recently, another study discussed this poor specificity and generated new variants using phage display with higher specificity.10
Activity-based probes (ABPs) based on the ubiquitin structure are commonly used to determine the activity of DUBs.11 Although these tools can target a wide range of enzymes exhibiting the same catalytic activity, our goal is to tailor them towards recognition of a specific target, to study it in detail. To achieve this selectivity, the structure of ubiquitin can be altered by creating mutations in certain regions of ubiquitin as mentioned before. Here, we used several methods to generate an activity-based probe for USP7 based on the structure of Ub. Phage display data, computational predictions and structural analysis were combined to enable the introduction of the desired selectivity. From more than 125 Ub mutants and various rounds of selection, we identified a mutant M6 from Library III (referred to as LIIIM6, see Scheme 1), showing mainly reactivity towards USP7. This allows us to study the activity of an individual DUB in a complex pool of enzymes with a highly-conserved catalytic structure. Moreover, the reasoning and the methodologies used can serve to establish a convenient approach for other reagent designs and additional interesting DUBs as well.
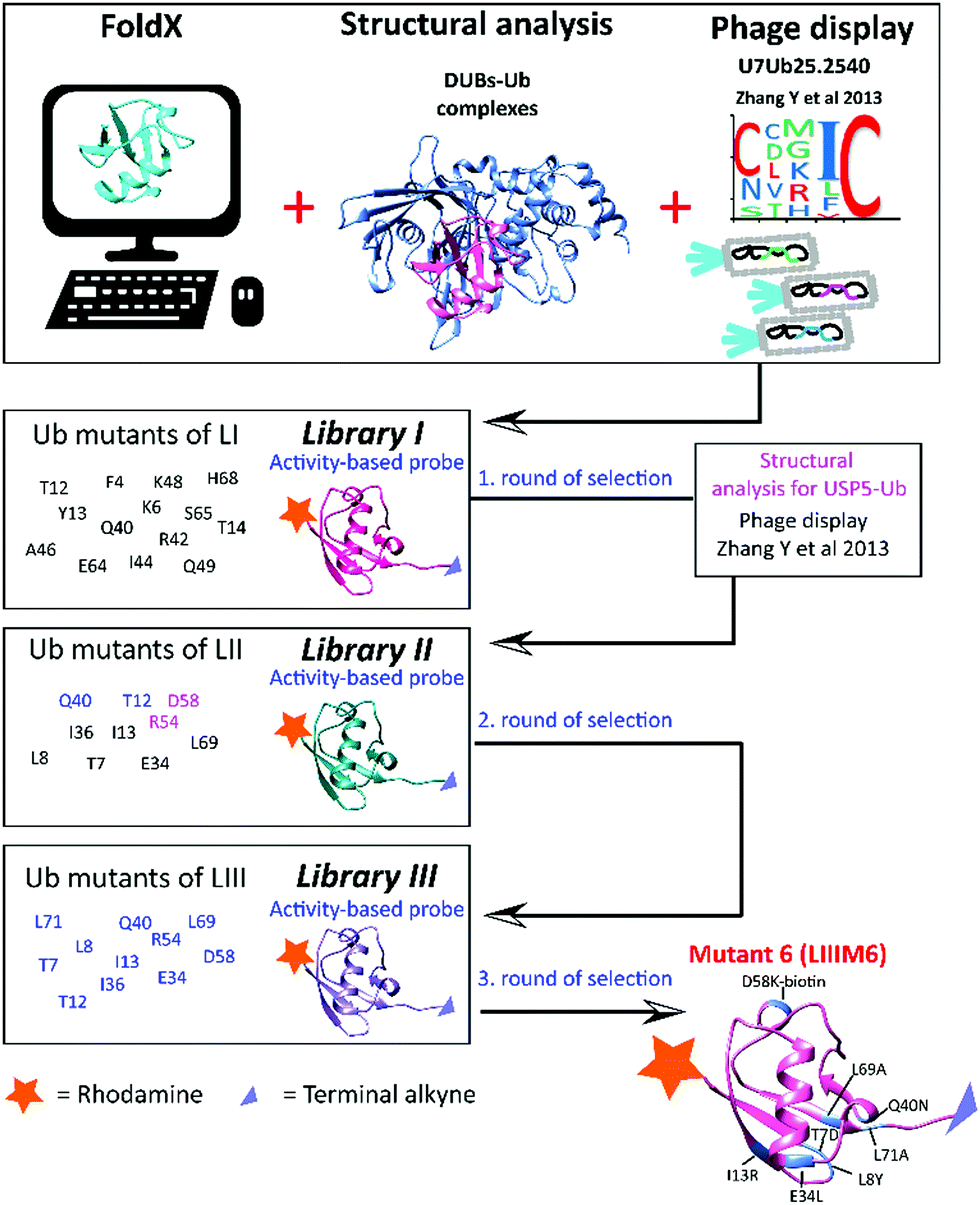 | ||
| Scheme 1 Schematic representation of the tools used in the library design and the selection process. The main mutations introduced in each of the libraries are shown on the left side of the scheme. | ||
Based on our previous knowledge in designing ABPs,12 we decided to integrate various mutations in the ubiquitin sequence in order to achieve high affinity towards USP7. We aimed to develop a reagent that reacts in a covalent fashion. This covalent interaction between probe and enzyme enables easy monitoring of the levels of our target during screenings of mutant libraries. Our strategy was to combine phage display data and structure-based computational mutational analysis using FoldX software.13 Phage display has proven to be a fast route towards enzyme-selective Ub mutants.7 These mutations can act as a starting point in the screening process. Additionally, the FoldX algorithm calculates the relative binding energies of the enzyme with ubiquitin containing in silico mutated amino acids, thus allowing the identification of residues that potentially optimize the interface between ubiquitin and USP7. Computational predictions were obtained not only for the USP7–Ub structure (PDB ID: 1NBF), but also for structures of ubiquitin with a few of the most abundant DUBs, including USP5 (PDB ID: 3IHP), USP14 (PDB ID: 2AYO), USP21 (PDB ID: 2Y5B) and UCHL3 (PDB ID: 1XD3). Results of the FoldX calculation for each of these DUB–Ub complexes are shown in the Supporting data 1 (ESI†). As anticipated, for many of the positions in the ubiquitin sequence the mutations would have similar effects as for the DUBs mentioned above. Therefore, those mutations in the ubiquitin sequence that were predicted to increase the binding only to USP7 and negatively impacting other DUB–Ub interactions were chosen as the most interesting ones. In the last rounds of improvements, we considered mutations that should exhibit a negative effect on USP5 based on structural analysis. These data were applied in designing the libraries by modifying the ubiquitin structure mainly through single or multiple point mutations (Scheme 1). The ubiquitin reagents were synthesized using Fmoc-based solid phase peptide synthesis as previously described.12 All synthetic ubiquitin mutants were modified at the N-terminus with a fluorescent label such as rhodamine or tamra using peptide coupling conditions. The protected ubiquitin molecule was cleaved from the trityl resin using a mixture of 20% (v/v) hexafluoroisopropanol in methylenechloride. Next, propargylamine was coupled to the C-terminus of Gly75 via standard peptide coupling, followed by global deprotection and purification.12 The terminal alkyne allows for a covalent reaction with the active-site cysteine of the target enzyme, while the fluorescent label enables an assay read-out. These types of reagents can be generated fully synthetically in parallel fashion, offering us flexibility and simplicity. Tests were performed with HAP1 cell lysates, which were incubated with the reagents for 30 min, followed by separation on SDS-PAGE and analysis of fluorescence. The labelling pattern of these types of reagents was established previously.12 For every ubiquitin variant, we interpreted the effect of each specific mutation, specifically on the decrease in labelling of other DUBs, compared to the wild-type reagent. Each round of synthesis yielded a separate group of mutants referred to by us as Library I (LI) through Library III (LIII) and each such library consists of several separate (i.e. unrelated) mutants referred to as M1, M2, etc. Various rounds of screening were performed, investigating different mutations obtained from the above mentioned tools leading to the most selective reagent.
In the first round of library design we considered the mutations present in the Ub variant U7Ub25.2540 (first example of a strong binder for USP7 as previously mentioned)9 and mutations derived from the FoldX data. In addition, due to the fact that the crystal structure of this Ub variant is available (PDB ID: 4HK2), the generated model in complex with USP7 was taken along in our computational efforts. We combined this scaffold with other point mutations from FoldX predictions. Various mutations were introduced to negatively impact other DUBs such as USP14 (e.g. F4W, T12W, and I36R) and UCHL3 (e.g. I36N, Q40N, I44K). Taken together, all these mutations were implemented in Library I (LI). Almost no reactivity was observed by covalent reagents that were designed based on the published phage display data. A few observations could be made based on the screening of LI (Fig. S1, ESI†), such as a decrease in USP14 labelling upon introduction of T12Q/W and T14I. In addition, Q40N showed a clear reduction in the labelling of DUBs in the low molecular weight region (Fig. S1b, ESI†), while still maintaining a similar level of labelled USP7.
The information acquired from our mutants (e.g. T12W, Q40N) was applied in the following library design referred as LII with subsequent addition of two new criteria: first reducing the affinity of the mutants towards USP5 and second other mutants selected from phage-display data.9 USP5, is one of the most abundant DUBs, and generally strongly labelled by most mutants. In Library II new synthetic ubiquitin mutants were included based on published phage display variants, along with predicted single point mutations to disfavour the binding towards USP5. In order to evaluate and alter the affinity of wild-type ubiquitin towards USP5, we analysed available structures of USP5 and USP7, investigating structural differences between these USPs and the types of interactions involved in the binding with ubiquitin. Based on this analysis, we predicted that the presence of a bulky group (such as tryptophan and lysine–biotin) on position 54 or 58 of ubiquitin would lead to steric hindrance with a specific region of USP5, which is absent in the structure of USP7 (see Videos S1, S2 and Fig S2, ESI†). This would then disrupt the interactions between USP5 with ubiquitin, potentially causing a decrease in affinity for USP5 with almost no consequences for binding to USP7. The translation of Ub variants that were identified during earlier phage display studies as strong binders to activity-based reagents proved not as straightforward. The reactivity as covalent probe depends on the proper alignment of a warhead to react and this is hard to predict prior to testing. As a result of introducing mutations, the structure might change in such a way that the variant may not be able to react with the active site cysteine residue. Based on the phage display data discussed above, we selected the mutations present in the Ub variants, which were matured to, U7Ub25.2540. The selected residues are in positions 7, 8, 13, 34, 36, 69 and 71.9 Of almost 40 members of LII, LIIM6 (T7D, L8Y, I13R, E34L, L69A, L71A) and LIIM7 (T7S, L8Y, I13Y, E34I, I36N, L69I, L71G) showed promise, mainly labelling DUBs in the high molecular weight region of the gel, including USP5 and USP7 (Fig. S3, ESI†). Notably, similar mutants derived from the phage display data showed no reactivity. This demonstrates the challenge in translating information from phage display data into active-site reactive probes. Based on the sequence of LIIM6 and LIIM7, we introduced single or combined mutations mainly in positions 12, 40, 54 and 58 to be evaluated in Library III. Fig. S4 (ESI†) shows the DUB activity assay for LIII, in which a few rhodamine-tagged mutant Ub probes demonstrated preference for labelling of USP7. The changes introduced in the sequence of the wild-type reagents led to an increase in USP7-selectivity, despite the presence of many other enzymes, which are structurally very similar to USP7. For the best mutants (shown in red, Fig. S4, ESI†), the labelling experiment was repeated at a lower concentration. The levels of labelled USP7 were also determined by western blotting. M6 from Library III (LIIIM6; T7D, L8Y, I13R, E34L, Q40N, D58K-biotin, L69A, L71A) demonstrated the highest reactivity compared to the other mutants of LIII (Fig. S5, ESI†). From now on M6 refers to LIIIM6. In Fig. 1, we show the labelling of HAP1 WT and USP7KO (knockout from HAP1 cells) lysate with wild-type or M6 ubiquitin probe, using both gel-based fluorescence analysis (using the N-terminally rhodamine-labelled probes), and western blotting for USP7.
 | ||
| Fig. 1 DUB activity-based probe assay of HAP1 and USP7 KO HAP1 lysate with Rh-M6-PA (M6, Library III) and Rh-UbWt-PA (WT). Labelled DUBs were analysed using in-gel fluorescence scanning and western blotting. β-Actin is used as loading control. * = labelled USP7. | ||
More recently, additional ubiquitin variants for USP7 from phage display data10 were reported (UbV.7.1–UbV.7.3), mutated at different positions compared to the previous data.9 These new mutations were incorporated in another round of screening and selection, individually as well as combined with the mutations in M6 (Fig. S6, ESI†). Unfortunately, translating the information acquired from this set of phage display data did not improve selectivity of M6 for USP7 further and for some of these mutants very low or almost no labelling of USP7 was observed. This demonstrates that phage-display is not good at predicting covalent reagents and emphasizes that a combination of more tools is necessary to reach the desired selectivity. In this elaborate screening and design process we identified M6 from more than 125 synthetic mutants, based on the combination of data coming from aforementioned efforts. We have designed a modified probe with a biotin handle (lysine–biotin in position 58) for pull-down experiments and a fluorescent tag for in-gel detection, which can covalently bind USP7 with high selectivity. To validate its selectivity and reactivity towards the pool of DUBs present in HAP1 cells, we performed pull-down experiments and further analysed selectivity by mass spectrometry (MS). ABPs have been used previously as chemoproteomics tools.14,15 As anticipated, Rh-M6-PA reacted with USP7, which was present in the pull-down as confirmed by western blotting for USP7. Fig. 2 and Fig. S7, Supplementary data 2 (ESI†) show the DUBs identified with LC-MS/MS analysis. From a cellular pool of proteins, USP7 was identified together with only two other DUBs, USP15 and USP16, and two known USP7 interactors (PPlL416 and DHX4017) to be significantly enriched, demonstrating high selectivity of the probe towards USP7.
 | ||
| Fig. 2 Pull-down experiments workflow with Rh-M6-PA and identification of the proteins using mass spectrometry. (a) Schematic representation of the pull-down experiments with Rh-M6-PA; (b) DUB activity-based probe assay of HAP1 and USP7KO HAP1 lysate with Rh-M6-PA. Labelled DUBs were analysed using in-gel fluorescence scanning and western blotting; (c) representation of the DUBs and USP7 interactors identified in the experiments. See also Supporting data 2 (ESI†). β-Actin is used as loading control. Control = HAP1 WT lysate incubated only with neutravidin beads. | ||
In conclusion, we showed the potential of combining various tools to design an activity-based probe with high selectivity for a target. This USP7-selective ubiquitin-based ABP is the first of its kind, allowing us to selectively determine active levels of USP7, and investigate cellular processes that depend on the activity of USP7. The information and the data already known for USP7 from phage display could not easily be employed to generate modifications to the sequence of ubiquitin to turn it into an active ABP, which may be due to misalignment of the C-terminus of the phage display Ub variants into the active site. The methodology described above was successfully applied for USP7 and can be used to design similar probes for other DUBs. Mutating the primary structure of ubiquitin or a ubiquitin-like protein can be further used to construct probes and in the future selective substrates, to study in real-time the catalytic activity of ubiquitin and ubiquitin-like proteases.
We would like to thank Dris El Atmioui for peptide synthesis, Patrick Celie for protein production and Farid El Oualid for suggestions. Work in the H. O. lab was supported by a Netherlands Organization for Scientific Research VICI grant (724.013.002) and in the A. C. O. V. lab by the ERC grant 310913. H. O. has been part of the DUB Alliance that includes Cancer Research Technology and FORMA Therapeutics that supported this work financially.
Conflicts of interest
H. O. is co-founder and shareholder of the company UbiQ.References
- D. Komander and M. Rape, Annu. Rev. Biochem., 2012, 81, 203–229 CrossRef CAS PubMed.
- A. Hershko and A. Ciechanover, Annu. Rev. Biochem., 1998, 67, 425–479 CrossRef CAS PubMed.
- D. Komander, M. J. Clague and S. Urbe, Nat. Rev. Mol. Cell Biol., 2009, 10, 550–563 CrossRef CAS PubMed.
- S. A. Abdul Rehman, Y. A. Kristariyanto, S. Y. Choi, P. J. Nkosi, S. Weidlich, K. Labib, K. Hofmann and Y. Kulathu, Mol. Cell, 2016, 63, 146–155 CrossRef CAS PubMed.
- B. Nicholson and K. G. Suresh Kumar, Cell Biochem. Biophys., 2011, 60, 61–68 CrossRef CAS PubMed.
- J. Zhou, J. Wang, C. Chen, H. Yuan, X. Wen and H. Sun, Med. Chem., 2018, 14, 3–18 CAS.
- A. Ernst, G. Avvakumov, J. Tong, Y. Fan, Y. Zhao, P. Alberts, A. Persaud, J. R. Walker, A. M. Neculai, D. Neculai, A. Vorobyov, P. Garg, L. Beatty, P. K. Chan, Y. C. Juang, M. C. Landry, C. Yeh, E. Zeqiraj, K. Karamboulas, A. Allali-Hassani, M. Vedadi, M. Tyers, J. Moffat, F. Sicheri, L. Pelletier, D. Durocher, B. Raught, D. Rotin, J. Yang, M. F. Moran, S. Dhe-Paganon and S. S. Sidhu, Science, 2013, 339, 590–595 CrossRef CAS PubMed.
- M. Gabrielsen, L. Buetow, M. A. Nakasone, S. F. Ahmed, G. J. Sibbet, B. O. Smith, W. Zhang, S. S. Sidhu and D. T. Huang, Mol. Cell, 2017, 68, 456–470 CrossRef CAS PubMed.
- Y. Zhang, L. Zhou, L. Rouge, A. H. Phillips, C. Lam, P. Liu, W. Sandoval, E. Helgason, J. M. Murray, I. E. Wertz and J. E. Corn, Nat. Chem. Biol., 2013, 9, 51–58 CrossRef CAS PubMed.
- W. Zhang, M. A. Sartori, T. Makhnevych, K. E. Federowicz, X. Dong, L. Liu, S. Nim, A. Dong, J. Yang, Y. Li, D. Haddad, A. Ernst, D. Heerding, Y. Tong, J. Moffat and S. S. Sidhu, J. Mol. Biol., 2017, 429, 3546–3560 CrossRef CAS PubMed.
- D. S. Hameed, A. Sapmaz and H. Ovaa, Bioconjugate Chem., 2017, 28, 805–815 CrossRef CAS PubMed.
- R. Ekkebus, S. I. van Kasteren, Y. Kulathu, A. Scholten, I. Berlin, P. P. Geurink, A. de Jong, S. Goerdayal, J. Neefjes, A. J. Heck, D. Komander and H. Ovaa, J. Am. Chem. Soc., 2013, 135, 2867–2870 CrossRef CAS PubMed.
- J. Schymkowitz, J. Borg, F. Stricher, R. Nys, F. Rousseau and L. Serrano, Nucleic Acids Res., 2005, 33, W382–W388 CrossRef CAS PubMed.
- D. S. Hewings, J. Heideker, T. P. Ma, A. P. AhYoung, F. El Oualid, A. Amore, G. T. Costakes, D. Kirchhofer, B. Brasher, T. Pillow, N. Popovych, T. Maurer, C. Schwerdtfeger, W. F. Forrest, K. Yu, J. Flygare, M. Bogyo and I. E. Wertz, Nat. Commun., 2018, 9, 1162 CrossRef PubMed.
- A. Borodovsky, H. Ovaa, N. Kolli, T. Gan-Erdene, K. D. Wilkinson, H. L. Ploegh and B. M. Kessler, Chem. Biol., 2002, 9, 1149–1159 CrossRef CAS PubMed.
- M. E. Sowa, E. J. Bennett, S. P. Gygi and J. W. Harper, Cell, 2009, 138, 389–403 CrossRef CAS PubMed.
- A. Georges, E. Marcon, J. Greenblatt and L. Frappier, Sci. Rep., 2018, 8, 15833 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc00969h |
| This journal is © The Royal Society of Chemistry 2019 |