
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of chiral nine and twelve-membered cyclic polyamines from natural building blocks†
Thomas
Müntener
 a,
Fabienne
Thommen
a,
Daniel
Joss
a,
Jérémy
Kottelat
b,
Alessandro
Prescimone
a and
Daniel
Häussinger
*a
a,
Fabienne
Thommen
a,
Daniel
Joss
a,
Jérémy
Kottelat
b,
Alessandro
Prescimone
a and
Daniel
Häussinger
*a
aDepartment of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: daniel.haeussinger@unibas.ch
bSchool of Engineering and Architecture of Fribourg, Boulevard de Pérolles 80, 1705 Fribourg, Switzerland
First published on 27th March 2019
Abstract
A rational strategy for the facile and efficient cyclization of amino acid-based linear precursors forming nine and twelve-membered cyclic peptidomimetics is reported. The resulting chiral lactams can readily be reduced to substituted cyclic polyamine analogues of 1,4,7,10-tetraaza-cyclododecane (cyclen) and 1,4,7-triaza-cyclononane (TACN).
The pharmaceutical industry has been treating diseases by administering drugs based on small molecules for decades. Many small-molecule drugs, however, show only limited specificity for their designated targets. Biomimetic therapeutics can overcome the shortcomings of small-molecule drugs due to their capability of selectively recognizing specific targets in highly crowded cellular media. Signalling peptides have crucial roles in human physiology1 due to their high affinity towards specific cell receptors and consequently, linear peptides have been investigated as possible new drug molecules. Generally, peptides are considered poor drugs due to their low oral uptake, fast metabolization by enzymatic degradation and low cell membrane permeability.2 Nevertheless, many peptide drugs are on the market due to their low toxicity and superior specificity. Especially cyclic peptides have proven to be very successful as therapeutic drugs2,3 as most often they show enhanced biological activities compared to their linear analogues. The structural bias caused by the limited conformational flexibility offers favourable binding properties by reducing the entropic penalty upon binding.3 Furthermore, the lack of both C- and N-termini drastically increases the stability towards enzymatic hydrolysis. Finally, some cyclic peptides show cell membrane permeability due to specific structural features.4 Many of the hurdles associated with both linear and cyclic therapeutic peptides can be circumvented upon chemical modification of canonical amino acids within the polypeptide chain creating peptidomimetics.5 Access to cyclic peptide or peptide-like nine and twelve-membered medium sized rings is challenging. Cyclization of linear tripeptides is notoriously difficult as cyclization can only occur if all peptide bonds are in the less favourable cis configuration.6 Cyclization proceeds only in moderate to high yields for those linear sequences containing strong turn inducers (like proline or pseudoproline) or medium turn inducers (side-chain protected amino acids e.g. benzyl protected glutamic acid or serine).7 For the slightly larger family of twelve-membered cyclic tetrapeptides the same problem of bringing both reactive ends together arises. Most macrocyclic tetrapeptides8 contain at least one proline (or pseudoproline)9 residue, a mixture of D- and L-amino acids10 or unnatural amino acids11,12 to facilitate the turn. Molecularly imprinted cavities can aid the macrocyclization of strained tetrapeptides by enforcing turn conformations.13 In general, high dilution conditions are required to perform cyclization in solution to prevent oligomerizations. Pseudo dilution conditions can be obtained by the attachment of the linear precursor on the surface of an insoluble polymer14 allowing for increased concentrations and simplified experimental procedures. In contrast, cyclic peptidomimetics can be accessed by a whole variety of different chemical tools including macro lactamization, macro lactonization, nucleophilic substitutions and multicomponent reactions.8 Furthermore, various transition-metal-catalysed reactions offer fast and selective ring-closing reactions.15 However, such reactions may introduce structural elements (e.g. double bonds or triazole rings), not seen in cyclic peptides made from natural amino acids. Nevertheless, the increased flexibility gained by the introduction of structural elements mentioned above, is usually beneficial for the cyclization. On the other hand, natural cyclic tri- and tetra-peptides or closely related peptidomimetics are potential retrosynthetic precursors of 1,4,7,10-tetraaza-cyclododecane (cyclen) and 1,4,7-triaza-cyclononane (TACN) derivatives. Stereospecifically substituted chiral cyclen derivatives are commonly synthesized using nucleophilic ring-openings of N-protected aziridines.16–18
In contrast, TACN and substituted analogues are most commonly synthesized using nucleophilic ring closing of a linear precursor.19,20 Cyclen, TACN and their derivatives are widely used in the synthesis of transition metal chelators applied in radiopharmacology,21 magnetic resonance imagining22 and paramagnetic NMR spectroscopy.23 Here, we report a synthetic strategy towards nine and twelve-membered cyclic peptidomimetics based on amino acids or simple derivatives which can easily be converted to the corresponding cyclic polyamine derivatives of cyclen or TACN.
Initially, we searched for a rational synthesis for stereospecifically substituted cyclens omitting the statistical oligomerization of aziridines which is extremely tedious to scale. Retrosynthetic analysis of M4-cyclen revealed that the target compound could be directly synthesized by the reduction of cyclic tetraalanine. Traditionally, cyclic tetrapeptides are synthesized by an intramolecular ring closing reaction either on resin or under high dilution conditions in solution both minimizing the formation of oligomers. We synthesized linear tetraalanine using standard solution peptide coupling conditions and attempted the macrocyclization on a polymer support as well as in solution. However, purification and isolation of the desired cyclic tetraalanine proved to be difficult and the isolated yields were generally low (<5%). We concluded that the unfavoured cis conformation of the peptide bond required for a successful cyclization severely hinders the formation of the desired cyclic tetrapeptide. Furthermore, we observed that cyclic tetraalanine as well as linear tetraalanine are highly insoluble compounds limiting the range of applicable solvents. Therefore, an alternative strategy based on the formation of a bisaminal24 with tetraalaninol was investigated due to the increased solubility and flexibility of tetraalaninol compared to tetraalanine. Only in the case of glyoxal we observed formation of the corresponding bisaminal, but subsequent ring-closing proved to be impossible in our hands. We therefore tested a combined approach exploiting the benefits of both procedures investigated so far using a fast peptide coupling as ring-closing step as well as the increased flexibility gained by the replacement of amide bonds with amines. Our new synthesis of M4-cyclen (cf.Scheme 1) starts with the assembly of the linear tetramer 14 from commercially readily available (S)-alanine derivatives. We envisioned a convergent approach starting with the N-terminal protection of (S)-alaninol with Cbz followed by an IBX mediated oxidation in ethyl acetate/dimethyl sulfoxide under reflux.25 Optical purity (>99%) of the amino aldehyde 1 was determined by chiral HPLC (cf. ESI,† S69). The subsequent reductive amination of 3 with alanine tert-butyl ester proceeded in excellent yields without epimerization (cf. ESI,† S73). To avoid side-reactions during the second chain elongation step, from a dimer to the tetramer, the non-terminal amines were protected using benzylbromide in acetonitrile providing access to the corresponding dimer 5. Subsequent acidic hydrolysis or hydrogenolytic reduction of dimer 5 afford the deprotected dimer 12 or 9, respectively. However, we found that the Cbz deprotection could not be carried out selectively under all tested conditions using various activated and deactivated palladium-based catalysts, solvents, temperatures and sources of hydrogen. Therefore, we introduced the Alloc protecting group as an alternative which offers a third orthogonality and can selectively be deprotected in the presence of the benzyl group using phenylsilane and Pd(PPh3)4. After a HATU mediated peptide coupling between the dimers 9 and 12 followed by simultaneous removal of both the Cbz and tert-butyl protecting group under strongly acidic conditions using hydrobromic acid in acetic acid, the linear tetramer 14 was obtained as HBr salt. We found that best macrocyclization yields of up to 85% were obtained using HATU as peptide coupling reagent and a dilution of 1 g L−1 in acetonitrile. Remarkably, even at concentrations as low as 2 mM (1 g L−1) the formation of the active ester was fast and macrocyclization was completed within less than 30 minutes at room temperature and no larger oligomers were detected. However, we found that in order to avoid N-acetylation and N-benzylation side reactions, it was crucial to remove traces of acetic acid and benzyl bromide from the linear tetramer 14 HBr salt by prolonged drying under vacuum at 40 °C. Applying a trimethylsilyl chloride activated amide reduction26 with lithium aluminium hydride followed by a transfer hydrogenation affords the macrocyclic compound M4-cyclen in 15 steps with an overall yield of 22%.
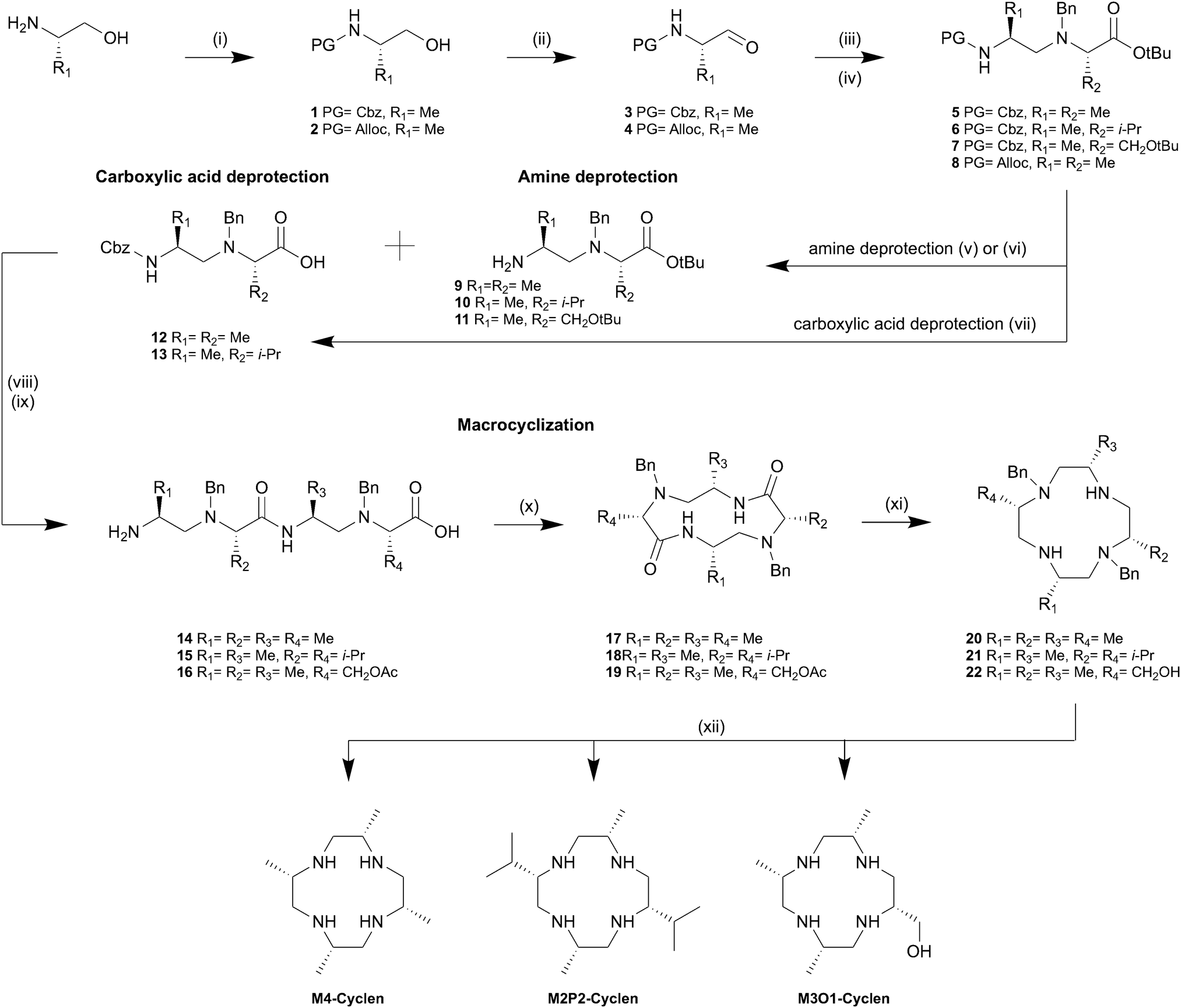 | ||
| Scheme 1 Syntheses of 2S,5S,8S,11S-2,5,8,11-tetra-methyl-cyclen (M4-cyclen), 2S,5S,8S,11S-2,8,-diisopropyl-5,11-di-methyl-cyclen (M2P2-cyclen) and 2S,5S,8S,11S-2-hydroxymethyl-5,8,11-tri-methyl-cyclen (M3O1-cyclen). Reagents and conditions: (i) Cbz-Cl or Alloc-Cl, Na2CO3, EtOAc/H2O, RT, 1 h; (ii); IBX, DMSO, EtOAc, reflux, 2–5 h (iii) amine, NaBH(OAc)3, CH2Cl2, 20–25 °C, 2–16 h; (iv) BnBr, K2CO3, MeCN, 40–50 °C, 8–16 h; (v) PhSiH, Pd(PPh3)4, CH2Cl2, 20–25 °C, 4 h; (vi) Pd/BaSO4, H2, MeOH, 20–25 °C, 4–16 h; (vii) HCl in dioxane, 40 °C, 16 h; (viii) HATU, DIPEA, MeCN, 20–25 °C, 2–4 h; (ix) HBr in acetic acid 16 wt%, 40 °C, 30 min; (x) HATU, DIPEA, MeCN, 20–25 °C, 20 min; (xi) TMS-Cl, LiAlH4, THF/CH2Cl2, 0–5 °C → 20–25 °C, 3 h; (xii) Pd/C, ammonium formate, EtOH, reflux, 16 h or Pd/C, H2, MeOH, RT, 16 h. | ||
We tested the general applicability of this synthetic strategy and synthesized two additional chiral cyclen derivatives with different substitution patterns and functional groups. M2P2-cyclen (C2-symmetric) is accessible by the replacement of alanine tert-butyl ester with its valine analogue during the reductive amination step. Similarly, M3O1-cyclen is obtained by replacing the dimer 9 with 11 prior to the second elongation step. Surprisingly we found that the Cbz protecting group can be selectively removed from dimers 6 and 7 using deactivated Pd on BaSO4 under 1 atmosphere of hydrogen omitting the need for an additional protecting group. We attribute this finding to the increased steric shielding of the N-benzyl protecting group by the larger substituents, causing an increased selectivity towards the terminal Cbz protecting group. Therefore, M2P2-cyclen can be synthesized in a convergent fashion in 11 steps with an overall yield of 24%. The asymmetric M3O1-cyclen was obtained in 13 steps with an overall yield of 17%. The larger substituents proved to be beneficial for the selective Cbz deprotection, however, we observed a decrease in reaction rates and lower yields for the macrocyclization step. Unfortunately, in addition we observed epimerization during the reductive amination in both cases leading to approximately 8% of unwanted diastereomers (cf. ESI,† S72). Nevertheless, we were able to grow single crystals suitable for X-ray analysis for all three twelve-membered macrocycles which in combination with NMR-spectroscopy prove the correct overall stereochemistry of the final product. We found that unwanted diastereomers formed during the reductive amination are gradually removed in the following steps. In particular, we observed a significant decrease of the unwanted diastereomers after the macrocyclization step. We hypothesize that the unwanted diastereomers have a less favourable pre-organization for cyclization. Furthermore, we observed that the cyclic unwanted diastereomers were easily separated by flash column chromatography at this stage. Inspired by the broad applicability of this synthetic strategy we challenged our method by applying it to smaller nine-membered triaza cycles. We envisioned the linear trimer 27 as the key molecule which should allow cyclization in a similar manner as for its larger twelve-membered analogue (cf.Scheme 2). The elongation of dimer 24 by one subunit was achieved by Cbz deprotection of dimer 23 followed by a second reductive amination step with the aldehyde 3, which exclusively favoured the primary amine over the secondary amine. The secondary amines were subsequently benzyl protected and deprotection of the C- and N-termini was achieved quantitatively using hydrobromic acid in acetic acid. Cyclization of the trimer 27 under identical conditions (1 g L−1 and HATU) yielded the benzyl protected lactam 28 in 63% yield. Although the first reductive amination leading to dimer 23 does not lead to epimerization, the second reductive amination causes epimerization to a similar extend as for M2P2 and M3O1. Similar to the findings presented for the twelve-membered macrocycles the unwanted diastereomers could be removed quantitatively by flash column chromatography of lactam 28. The subsequent reduction and transfer hydrogenation steps were carried out accordingly. Overall, M3-TACN is accessible in 10 steps in 19% overall yield.
 | ||
| Scheme 2 Synthesis of M3TACN. Reagents and conditions: (i) Pd/C, H2, MeOH, 20–25 °C, 4 h; (ii) NaBH(OAc)3, CH2Cl2, 20–25 °C, 16 h; (iii) BnBr, K2CO3, MeCN, 20–25 °C, 16 h; (iv) HBr in acetic acid 16 wt%, 40 °C, 30 min; (v) HATU, DIPEA, MeCN, 20–25 °C, 30 min; (vi) TMS-Cl, LiAlH4, THF/CH2Cl2, 0–5 °C → 20–25 °C, 5 h; (vii) Pd/C, ammonium formate, EtOH, reflux, 16 h. | ||
In conclusion we developed a new rational synthetic strategy for the cyclization of linear amino acid-based precursors giving accesses to new cyclic peptidomimetics consisting of nine-membered lactams and twelve-membered bislactams. Due to the modular build-up of the linear cyclization precursor a large number of different substitution patterns are accessible giving rise to novel symmetric and asymmetric cyclic lactams. The macrocyclization occurs in solution under moderate dilution conditions with high efficiency and yields. We anticipated that our method could find application in the synthesis of novel cyclic peptidomimetics with potential biological activities. Furthermore, the synthesis also provides access to new cyclen and TACN based derivatives for future application as high affinity metal chelators in paramagnetic NMR spectroscopy, magnetic resonance imaging or radiopharmacology. We are currently exploring new lanthanide chelating tags based on substituted cyclen derivatives.
The authors gratefully acknowledge financial support for this work from the Fondation Claude et Giuliana, Vaduz, Liechtenstein and the Department of Chemistry, University of Basel, Switzerland. We are grateful to R. Witzig for help with chiral HPLC analyses. We are indebted to C. E. Housecroft, E. C. Constable and Ch. Sparr for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed.
- S. H. Joo, Biomol. Ther., 2012, 20, 19–26 CrossRef CAS PubMed.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- Y. U. Kwon and T. Kodadek, Chem. Biol., 2007, 14, 671–677 CrossRef CAS PubMed.
- N. Qvit, S. J. S. Rubin, T. J. Urban, D. Mochly-Rosen and E. R. Gross, Drug Discovery Today, 2017, 22, 454–462 CrossRef CAS.
- G. Kartha, G. Ambady and P. V. Shanker, Nature, 1974, 247, 204–205 CrossRef CAS.
- S. Chakraborty, S. H. Lin, D. Shiuan and D. F. Tai, Amino Acids, 2015, 47, 1495–1505 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed.
- K. A. Fairweather, N. Sayyadi, I. J. Luck, J. K. Clegg and K. A. Jolliffe, Org. Lett., 2010, 12, 3136–3139 CrossRef CAS PubMed.
- S. F. Brady, S. L. Varga, R. M. Freidinger, D. A. Schwenk, M. Mendlowski, F. W. Holly and D. F. Veber, J. Org. Chem., 1979, 44, 3101–3105 CrossRef CAS.
- B. K. W. Chung, C. J. White, C. C. G. Scully and A. K. Yudin, Chem. Sci., 2016, 7, 6662–6668 RSC.
- M. P. Glenn, M. J. Kelso, J. D. Tyndall and D. P. Fairlie, J. Am. Chem. Soc., 2003, 125, 640–641 CrossRef CAS PubMed.
- D. F. Tai and Y. F. Lin, Chem. Commun., 2008, 5598–5600, 10.1039/b813439a.
- J. S. Davies, J. Pept. Sci., 2003, 9, 471–501 CrossRef CAS PubMed.
- L. R. Malins, Curr. Opin. Chem. Biol., 2018, 46, 25–32 CrossRef CAS.
- R. S. Ranganathan, R. K. Pillai, N. Raju, H. Fan, H. Nguyen, M. F. Tweedle, J. F. Desreux and V. Jacques, Inorg. Chem., 2002, 41, 6846–6855 CrossRef CAS PubMed.
- L. Dai, C. M. Jones, W. T. K. Chan, T. A. Pham, X. Ling, E. M. Gale, N. J. Rotile, W. C. Tai, C. J. Anderson, P. Caravan and G. L. Law, Nat. Commun., 2018, 9, 857 CrossRef PubMed.
- S. Kamioka, T. Takahashi, S. Kawauchi, H. Adachi, Y. Mori, K. Fujii, H. Uekusa and T. Doi, Org. Lett., 2009, 11, 2289–2292 CrossRef CAS PubMed.
- P. Désogère, Y. Rousselin, S. Poty, C. Bernhard, C. Goze, F. Boschetti and F. Denat, Eur. J. Org. Chem., 2014, 7831–7838 CrossRef.
- Y. Huang, Y. J. Liu, S. Liu, R. B. Wu and Z. H. Wu, Eur. J. Org. Chem., 2018, 1546–1551 CrossRef CAS.
- N. Wu, C. S. Kang, I. Sin, S. Ren, D. Liu, V. C. Ruthengael, M. R. Lewis and H. S. Chong, J. Biol. Inorg. Chem., 2016, 21, 177–184 CrossRef CAS PubMed.
- K. Jaudzems, X. Jia, H. Yagi, D. Zhulenkovs, B. Graham, G. Otting and E. Liepinsh, J. Mol. Biol., 2012, 424, 42–53 CrossRef CAS PubMed.
- C. Nitsche and G. Otting, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 98–99, 20–49 CrossRef CAS PubMed.
- A. Prokhorov, N. Le Bris, H. Bernard, G. Claudon and H. Handel, Synth. Commun., 2006, 36, 3271–3282 CrossRef CAS.
- J. D. More and N. S. Finney, Org. Lett., 2002, 4, 3001–3003 CrossRef CAS.
- B. Ravinder, S. R. Reddy, A. P. Reddy and R. Bandichhora, Tetrahedron Lett., 2013, 54, 4908–4913 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, analytical data and crystallographic data. CCDC 1886728–1886730. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc00720b |
| This journal is © The Royal Society of Chemistry 2019 |