
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A bio-inspired imidazole-functionalised copper cage complex†
Sarah C.
Bete
,
Christian
Würtele
and
Matthias
Otte
 *
*
Institut für Anorganische Chemie, Universität Göttingen, Tammannstraße 4, 37077 Göttingen, Germany. E-mail: matthias.otte@chemie.uni-goettingen.de
First published on 27th March 2019
Abstract
An imidazole-functionalised cage is synthesised that can coordinate to Cu(I). X-ray analysis reveals a T-shaped coordination of copper by the imidazole ligands reminiscent of the coordination geometry found in enzymatic active sites. This cage complex can catalyse the oxidation of benzylic alcohols to benzaldehydes utilizing oxygen as the terminal oxidant.
The coordination of imidazole moieties from histidines to copper ions plays a key role in many enzymatic active sites. Examples are particulate methane monooxygenases (pMMO),1,2 lytic polysaccharide monooxygenases (LPMO)1 and tyrosinases (Ty, Fig. 1a–c).3 These offer fascinating reactivities and selectivities that can often not be achieved by man-made catalysts. Chemists are inspired to develop (functional) molecular compounds that mimic these active sites. This mimicking has mostly been devoted towards the first coordination sphere, resulting in coordination complexes of rather low molecular weight.4–7
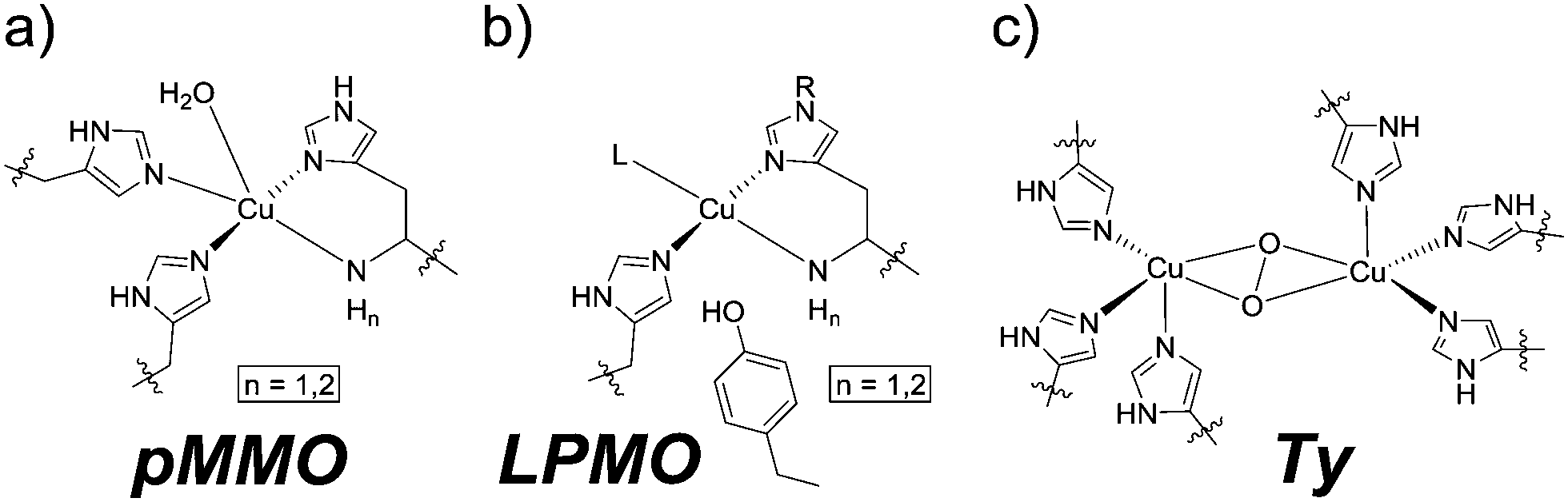 | ||
| Fig. 1 Active sites of (a) pMMO, (b) LPMO and (c) Ty. | ||
In comparison, the design of cavity-based metal complexes has received way less attention.8,9 This approach may allow to mimic also features of the active site pocket. It may also grant the advantages of chemistry in synthetic cages, such as protection of the catalytic active centre, stabilization of highly reactive species, control of binding events and unusual selectivities.10 To date, the mimicking of enzymatic active sites containing histidine by imidazole-functionalised cavity based ligands is mainly dominated by cyclodextrins and calix[6]arenes where the metal is actually located outside of the cavity with its binding site pointing towards it.11 Notably, it has been demonstrated very recently that imidazole-functionalised metal organic frameworks are able to coordinate to copper resulting in active catalysts for the conversion of methane to methanol.12
While active sites of enzymes containing metalloporphyrins have been mimicked by the use of molecular cage compounds,13 examples of molecular cages that offer such a well-defined ligand sphere to mimic non-heme enzymatic active sites via imidazole coordination are yet unprecedented. This is surprising as endo-functionalised organic cages have been shown to bind catalytic active centers.14 Costas and Ribas and co-worker recently synthesised Zn-, Cu- and Fe-coordination complexes based on pyridine coordination within a self-assembled nanocage.15 However, these species have not been reported to be catalytically active for the transformation of organic substrates.
Here we present the synthesis and characterization of the aza-cyclophane 1 (Scheme 1) and show that it is able to coordinate to copper(I) resulting in a catalyst for the oxidation of benzylic alcohols. Cage 1 was synthesised from known 2 (Scheme 1). Via nucleophilic substitution with an imidazolium salt, 3 was obtained in a yield of 54%. Afterwards, a Suzuki–Miyaura-coupling gave the imidazole-functionalised building block 4 in 52% yield. Single crystals suitable for X-ray diffraction analysis were obtained by layer diffusion of pentane into a dichloromethane solution of 4. 4 and the known compound 5 were subject to a reductive amination protocol with the aim to obtain 1. Such an approach is well documented for the synthesis of organic cages.16 After stirring a CH2Cl2/CH3OH solution of 4 and 5 for 2 days at room temperature and subsequent addition of sodium borohydride and purification, cage 1 was isolated in 46% yield.
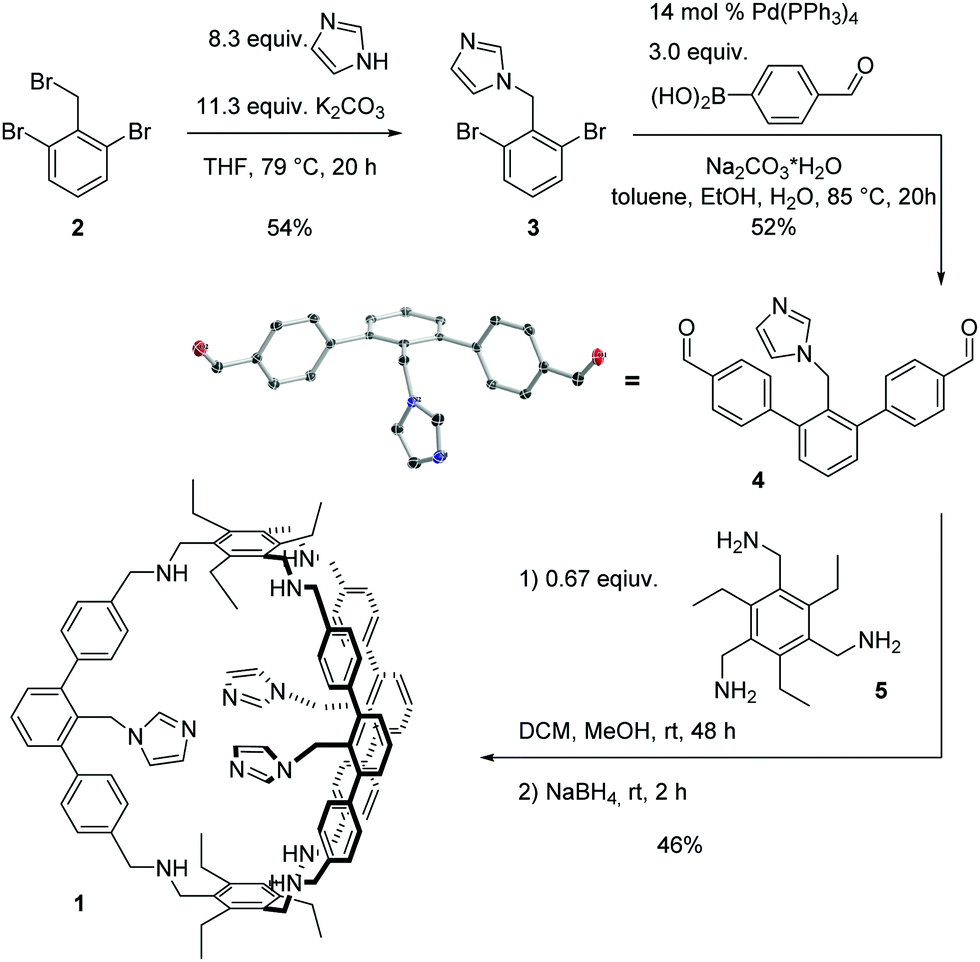 | ||
| Scheme 1 Synthesis of cage 1 and molecular structure of 4 in the solid state. | ||
1 was characterised via ESI-MS, 1H, 1H DOSY, 13C NMR and IR spectroscopy. The ESI-MS reveals signals with m/z values of 1501.8892, 751.4488 and 501.3016 corresponding to 1 + H+, 1 + 2H+ and 1 + 3H+. In the 1H NMR spectrum, two singlets are found at chemical shifts of 3.90 and 3.88 ppm that indicate the presence of benzylic amines (Fig. 2a). Three signals with relative integrals of three hydrogens each corresponding to the hydrogens of the imidazoles are observed, showing the imidazole's equivalence on NMR-timescale. The 1H DOSY NMR spectrum (see ESI†) reveals only one molecule size apart from solvents and trace amounts of grease. A diffusion coefficient of 4.1 × 10−10 m2 s−1 is found in chloroform. Using the Stokes–Einstein equation for spherically shaped molecules, this diffusion coefficient translates to a hydrodynamic radius of 9.8 Å.
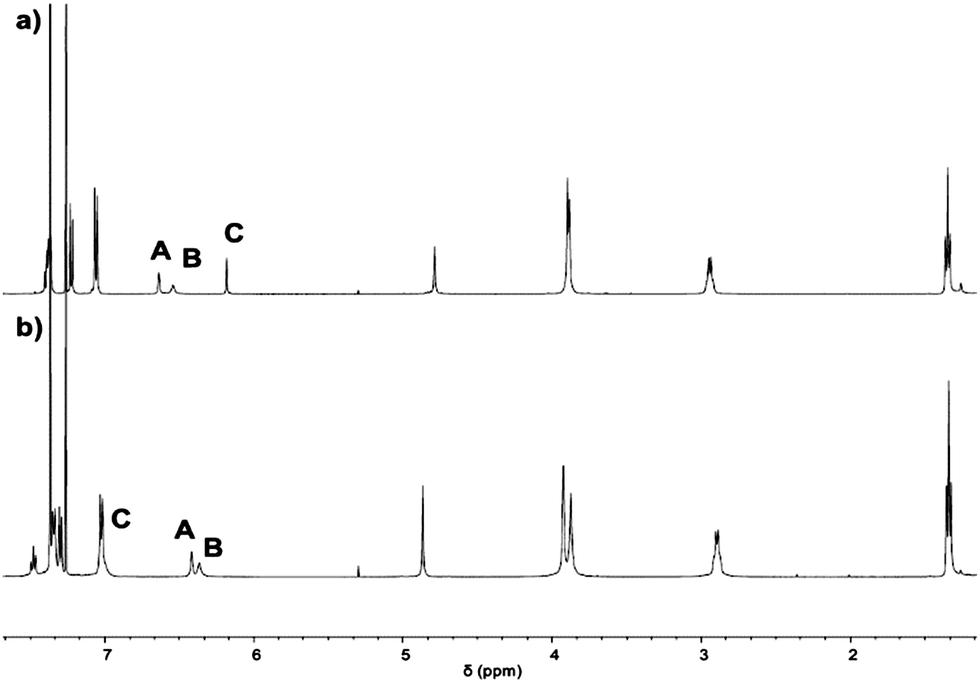 | ||
| Fig. 2 1H NMR spectra of (a) 1 and (b) [1-Cu]BF4 in CDCl3. | ||
With 1 in hand, we studied its behaviour to act as ligand for copper(I) (Scheme 2). Reaction with 0.9 equivalents Cu(MeCN)4BF4 (6) in dichloromethane at room temperature allows for the isolation of [1-Cu]BF4 as a white solid in 77% yield. The 1H NMR spectrum of [1-Cu]BF4 shows remarkable changes compared to the one of 1 (Fig. 2b). All three signals that are assigned to the imidazole protons (labelled with A–C) shift upon addition of 6. These shifts indicate that a coordination of the imidazole to copper was successful and that all imidazoles behave identically on NMR-timescale. Solutions of [1-Cu]BF4 turn greenish within minutes upon stirring under air (see ESI† for UV Vis spectra). The NMR spectrum of [1-Cu]BF4 changes significantly. No separated signals corresponding to the imidazole protons and the methylene-bridge could be assigned caused by broadening of all signals (see ESI†). A less pronounced broadening is observed for the backbone signals further indicating a coordination of copper to the imidazole moieties. The 1H DOSY NMR spectrum (see ESI†) of [1-Cu]BF4 in chloroform shows a single compound with a diffusion coefficient of 4.1 × 10−10 m2 s−1 which translates to a hydrodynamic radius of 9.9 Å. These values are close to the ones obtained from the 1H DOSY NMR spectrum of 1, which indicates that copper-coordination does not lead to any dimeric or oligomeric species. ESI-MS investigations of [1-Cu]BF4 shows species with m/z values of 1563.8112, 782.4088 and 1652.8288 corresponding to [1-Cu]+, [1-Cu]+ + H+ and [1-Cu]BF4 + H+.
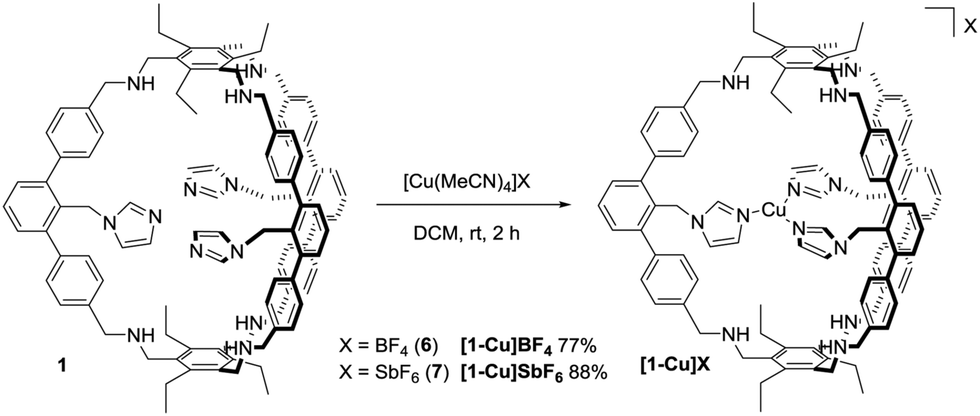 | ||
| Scheme 2 Synthesis of [1-Cu]BF4 and [1-Cu]SbF6. | ||
X-ray analysis of single crystals of [1-Cu]BF4, obtained by slow diffusion of THF into a diethylether solution, lead to a low data quality (see ESI†). However, the overall connectivity of the cage and its coordination to copper by all three imidazole units in the solid state could be confirmed. To obtain data of higher quality, we synthesised [1-Cu]SbF6 from 1 and Cu(MeCN)4SbF6 (7). NMR and MS gave analogous data compared to [1-Cu]BF4 (see ESI†). To our delight, we were able to obtain crystallographic data of higher quality by analysing single crystals of [1-Cu]SbF6 that form in a saturated ethereal solution at room temperature. As anticipated, copper is coordinated by each of the imidazole moieties (Fig. 3). A T-shaped coordination mode is found with N–Cu–N angles of 150°, 106° and 102°. Cu–N bond lengths of 1.9 Å are found for the imidazole units that are trans to each other, while the one for the remaining imidazole ligand is 2.1 Å. These values correspond well with the ones found in enzymatic active sites also showing a T-shaped coordination of the Cu(I) center. The Cu(I)–N(His) bond lengths in LPMOs are 1.9 Å, while the Cu(I)–NH2 bond length is 2.1 Å.1 The Cu(I)–N(His) bond lengths in the pMMO active site are 1.8 Å, 1.9 Å and 2.2 Å.17 The average distance between copper and the terminal carbon of the ethyl groups is 10 Å, which is in good agreement with the hydrodynamic radius obtained for [1-Cu]BF4. For the SbF6−-counterion three different locations are found (see ESI†) with the two most populated being located on the cage face with the largest N–Cu–N bond angle. The obtained crystallographic data shows four possible positions for two diethylether molecules (see ESI†). Three of these are inside the cage cavity. Hydrogen bonding interactions are observed between the cage N–H moieties and the ether and the SbF6− anion as acceptors.
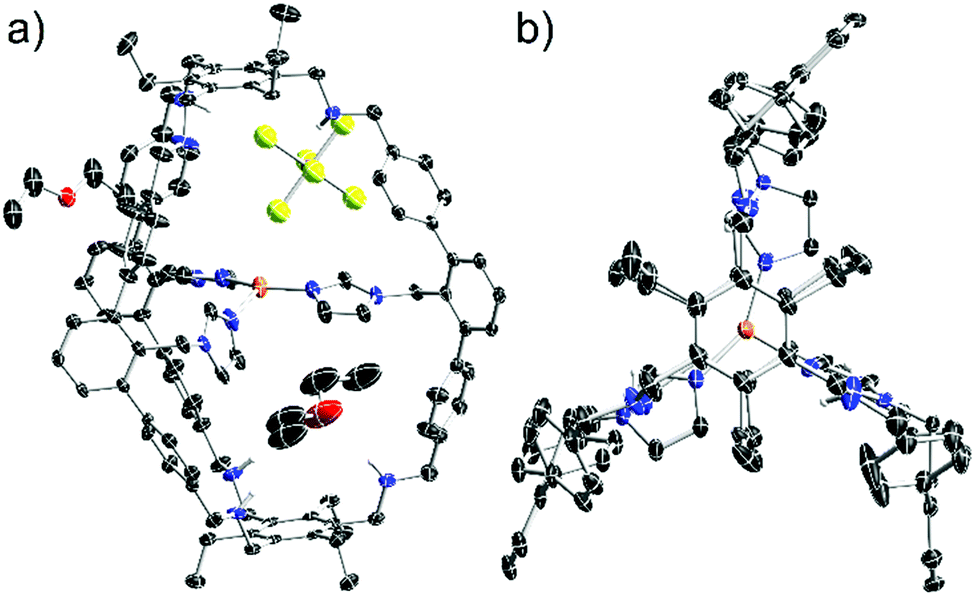 | ||
| Fig. 3 Molecular structure of [1-Cu]SbF6 in the solid state: (a) side view; (b) top view (solvent molecules and anion undisplayed). Carbon bonded hydrogen atoms are omitted for clarity. | ||
We next studied the ability of [1-Cu]BF4 to catalyse the oxidation of organic substrates. We chose the oxidation of 4-methoxybenzyl alcohol (8) to 4-anisaldehyde (9) with catalytic amounts of TEMPO (2,2,6,6-tetramethylpiperidin-1-yl)oxyl in a DCM/water mixture under aerobic conditions as model reaction (Table 1).18 Performing the reaction for 1.5 h resulted in 7 turnovers (entry 1), while overnight reaction time led to 11 turnovers (entry 2). Increasing [1-Cu]BF4 loading to 10 mol% resulted in 88% yield (entry 3), while increasing the substrate concentration to 0.50 M resulted in 28% yield (entry 4). Performing the reaction in absence of [1-Cu]BF4 gave only traces of 9 (entry 5). Control experiments with copper salt 6, empty cage 1 or without TEMPO showed no or only very little formation of 9 (maximum yield = 1%; entries 6 to 8). These results show that [1-Cu]BF4 is capable of oxidising organic substrates at room temperature in a catalytic fashion via employing oxygen as terminal oxidant. For the transformation of 8 to 9 the catalytic performance of [1-Cu]BF4 is comparable to other copper based systems. Using 6 in the presence of three equivalents of N-methylimidazole (NMI) gave 9 in 37% yield (entry 9),19 while Cu(MeCN)4OTf (10) with 2,2′-bipyridine (bipy) and N-methylimidazole gave 39% yield (entry 10).18
|
|
|||
|---|---|---|---|
| Entry | Catalyst ([mol%]) | Yield [%] | t [h] |
| a Substrate concentration = 0.11 M; all reactions under air; 7 mol% TEMPO; 11.1 equiv. H2O rt; all yields were determined by 1H NMR spectroscopy of the crude product mixtures in presence of internal standards. b Substrate concentration = 0.50 M. c Without TEMPO. NMI = N-methylimidazole. bipy = 2,2′-bipyridine. | |||
| 1 | [1-Cu]BF4 (1) | 7 | 1.5 |
| 2 | [1-Cu]BF4 (1) | 11 | 14 |
| 3 | [1-Cu]BF4 (10) | 88 | 16 |
| 4b | [1-Cu]BF4 (1) | 28 | 14 |
| 5 | None | <1 | 14 |
| 6 | 6 (1) | 1 | 14 |
| 7 | 1 (1) | 0 | 14 |
| 8c | [1-Cu]BF4 (1) | <1 | 14 |
| 9b | 6·3(NMI) (1) | 37 | 14 |
| 10b | 10·(bipy)·2(NMI) (1) | 39 | 14 |

We were wondering if [1-Cu]BF4 may have the ability to discriminate between substrates in competition experiments, especially compared to 6·3(NMI) and 10·(bipy)·2(NMI). When a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 3,5-di-methylbenzyl alcohol and 3,5-di-tert-butyl-benzyl alcohol was reacted with 1 mol% [1-Cu]BF4 and 7 mol% TEMPO the overall yield was 6% with a selectivity of 59:41 for the smaller substrate (Table 2, entry 1), whereas 6·3(NMI) preferably oxidises the larger 3,5-di-tert-butyl-benzyl alcohol with a selectivity of 42:58 (entry 2). 10·(bipy)·2(NMI) shows with 52:48 no preference (entry 3). Changing the smaller substrate to benzyl alcohol resulted in a comparable yield and a selectivity of 67:33 (entry 4). Again, the other two tested systems showed with 55:45 and 52:48 a smaller preference for the smaller substrate. Additional competition experiments of substrate pairs where the steric demand is further remote of the functional group and electronic differences are likely to be the cause of selectivity can be found in the ESI.† Although the effect is not too pronounced, these preliminary results show that there is a trend that [1-Cu]BF4 preferably catalyses the conversion of the smaller substrate.
:1 mixture of 3,5-di-methylbenzyl alcohol and 3,5-di-tert-butyl-benzyl alcohol was reacted with 1 mol% [1-Cu]BF4 and 7 mol% TEMPO the overall yield was 6% with a selectivity of 59:41 for the smaller substrate (Table 2, entry 1), whereas 6·3(NMI) preferably oxidises the larger 3,5-di-tert-butyl-benzyl alcohol with a selectivity of 42:58 (entry 2). 10·(bipy)·2(NMI) shows with 52:48 no preference (entry 3). Changing the smaller substrate to benzyl alcohol resulted in a comparable yield and a selectivity of 67:33 (entry 4). Again, the other two tested systems showed with 55:45 and 52:48 a smaller preference for the smaller substrate. Additional competition experiments of substrate pairs where the steric demand is further remote of the functional group and electronic differences are likely to be the cause of selectivity can be found in the ESI.† Although the effect is not too pronounced, these preliminary results show that there is a trend that [1-Cu]BF4 preferably catalyses the conversion of the smaller substrate.
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Cu] ([mol%]) | R1 | R2 |
PR1:PR2 |
Yield [%] |
| a Substrate concentration = 0.11 M; reactions under air; 7 mol% TEMPO; 11.1 equiv. H2O; rt; 5.0 h. All yields were determined by 1H NMR spectroscopy of the crude product mixtures in presence of internal standards. NMI = N-methylimidazole. bipy = 2,2′-bipyridine. | |||||
| 1 | [1-Cu]BF4 (1) | Me | t-Bu | 59:41 |
6 |
| 2 | 6·3(NMI) (1) | 42:58 |
22 | ||
| 3 | 10·(bipy)·2(NMI) (0.6) | 52:48 |
21 | ||
| 4 | [1-Cu]BF4 (1) | H | t-Bu | 67:33 |
7 |
| 5 | 6·3(NMI) (1) | 55:45 |
4 | ||
| 6 | 10·(bipy)·2(NMI) (0.6) | 52:48 |
3 | ||
In conclusion, we have synthesised an organic cage whose endo-functionalised cavity offers three imidazole units. These can coordinate to copper(I), assembling a motif as it is observed in the active sites of many oxygenases. This complex is the first example of an imidazole employing mimic of non-heme enzymatic active sites offering histidine coordination that is assembled in the cavity of a molecular cage. We used a protocol based on catalytic amounts of TEMPO for the catalytic oxidation of alcohols to aldehydes under aerobic conditions. The terminal oxidant in that protocol is oxygen. Preliminary results suggest that [1-Cu]BF4 preferably oxidises the smaller substrates in competition experiments. We believe that this trend can be improved significantly as 1 is easily accessible and therefore also easily altered.20 Derivatisation of 1 has great potential to give mimics of ligand spheres of other enzymatic active sites involving histidine coordination. Our aim with this highly tunable system is to design detailed structural mimics that beside having interesting reactivities for itselfs may lead to a better understanding of these enzymes and consequently facilitate the specific design of new synthetic catalysts with unprecedented reactivities.
M. O. thanks the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – OT 540/2-1 and the Fonds der Chemischen Industrie for funding. Prof. Dr Sven Schneider is highly acknowledged for his very generous support. We thank Prof. Dr Siegfried Schindler for the donation of Cu(MeCN)4SbF6.
Conflicts of interest
There are no conflicts to declare.Notes and references
- L. Ciano, G. J. Davies, W. B. Tolman and P. H. Walton, Nat. Catal., 2018, 1, 571 CrossRef.
- V. C.-C. Wang, S. Maji, P. P.-Y. Chen, H. K. Lee, S. S.-F. Yu and S. I. Chan, Chem. Rev., 2017, 117, 8574 CrossRef CAS.
- E. I. Solomon, D. E. Heppner, E. M. Johnston, J. W. Ginsbach, J. Cirera, M. Qayyum, M. T. Kieber-Emmons, C. H. Kjaergaard, R. G. Hadt and L. Tian, Chem. Rev., 2014, 114, 3659 CrossRef CAS.
- S. I. Chan, Y.-J. Lu, P. Nagababu, S. Maji, M.-C. Hung, M. M. Lee, I.-J. Hsu, P. D. Minh, J. C.-H. Lai, K. Y. Ng, S. Ramalingam, S. S.-F. Yu and M. K. Chan, Angew. Chem., Int. Ed., 2013, 52, 3731 CrossRef CAS.
- (a) A. Kunishita, M. Kubo, H. Sugimoto, T. Ogura, K. Sato, T. Takui and S. Itoh, J. Am. Chem. Soc., 2009, 131, 2788 CrossRef CAS; (b) C. Würtele, E. Gaoutchenova, K. Harms, M. C. Holthausen, J. Sundermeyer and S. Schindler, Angew. Chem., Int. Ed., 2006, 45, 3867 CrossRef PubMed; (c) M. Kumar, N. A. Dixon, A. C. Merkle, M. Zeller, N. Lehnert and E. T. Papish, Inorg. Chem., 2012, 51, 7004 CrossRef CAS PubMed.
- (a) C. F. Martens, R. J. M. Klein Gebbink, M. C. Feiters and R. J. M. Nolte, J. Am. Chem. Soc., 1994, 116, 5667 CrossRef CAS; (b) J. A. Halfen, S. Mahapatra, E. C. Wilkinson, S. Kaderli, V. G. Young Jr., L. Que Jr., A. D. Zuberbühler and W. B. Tolman, Science, 1996, 271, 1397 CrossRef CAS; (c) C. Würtele, O. Sander, V. Lutz, T. Waitz, F. Tuczek and S. Schindler, J. Am. Chem. Soc., 2009, 131, 7544 CrossRef; (d) I. Garcia-Bosch, A. Company, J. R. Frisch, M. Torrent-Sucarrat, M. Cardellach, I. Gamba, M. Güell, L. Casella, L. Que Jr., X. Ribas, J. M. Luis and M. Costas, Angew. Chem., Int. Ed., 2010, 49, 2406 CrossRef CAS; (e) C. Citek, C. T. Lyons, E. C. Wasinger and T. D. P. Stack, Nat. Chem., 2012, 4, 317 CrossRef CAS; (f) R. Cao, C. Saracini, J. W. Ginsbach, M. T. Kieber-Emmons, M. A. Siegler, E. I. Solomon, S. Fukuzumi and K. D. Karlin, J. Am. Chem. Soc., 2016, 138, 7055 CrossRef CAS; (g) V. E. Goswami, A. Walli, M. Förster, S. Dechert, S. Demeshko, M. C. Holthausen and F. Meyer, Chem. Sci., 2017, 8, 3031 RSC.
- (a) M. Rolff, J. Schottenheim, G. Peters and F. Tuczek, Angew. Chem., Int. Ed., 2010, 49, 6438 CrossRef CAS; (b) A. Hoffmann, C. Citek, S. Binder, A. Goos, M. Rübhausen, O. Troeppner, I. Ivanović-Burmazović, E. C. Wasinger, T. D. P. Stack and S. Herres-Pawlis, Angew. Chem., Int. Ed., 2013, 52, 5398 CrossRef CAS; (c) K. V. N. Esguerra, Y. Fall, L. Petitjean and J.-P. Lumb, J. Am. Chem. Soc., 2014, 136, 7662 CrossRef CAS; (d) A. Arnold, R. Metzinger and C. Limberg, Chem. – Eur. J., 2015, 21, 1198 CrossRef CAS PubMed.
- (a) S. Blanchard, L. Le Clainche, M.-N. Rager, B. Chansou, J.-P. Tuchagues, A. F. Duprat, Y. Le Mest and O. Reinaud, Angew. Chem., Int. Ed., 1998, 37, 2732 CrossRef CAS; (b) G. N. Di Francesco, A. Gaillard, I. Ghiviriga, K. A. Abboud and L. J. Murray, Inorg. Chem., 2014, 53, 4647 CrossRef CAS PubMed.
- J.-N. Rebilly, B. Colasson, O. Bistri, D. Over and O. Reinaud, Chem. Soc. Rev., 2015, 44, 467 RSC.
- (a) D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2007, 129, 2746 CrossRef CAS; (b) P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS; (c) M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251 CrossRef CAS PubMed; (d) S. Freye, J. Hey, A. Torras-Galán, D. Stalke, R. Herbst-Irmer, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2012, 51, 2191 CrossRef CAS PubMed; (e) Q. Zhang and K. Tiefenbacher, J. Am. Chem. Soc., 2013, 135, 16213 CrossRef CAS PubMed; (f) M. Otte, ACS Catal., 2016, 6, 6491 CrossRef CAS.
- (a) I. Tabushi and Y. Kuroda, J. Am. Chem. Soc., 1984, 106, 4580 CrossRef CAS; (b) A. Parrot, S. Collin, G. Bruylants and O. Reinaud, Chem. Sci., 2018, 9, 5479 RSC.
- J. Baek, B. Rungtaweevoranit, X. Pei, M. Park, S. C. Fakra, Y.-S. Liu, R. Matheu, S. A. Alshmimri, S. Alshehri, C. A. Trickett, G. A. Somorjai and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 18208 CrossRef CAS PubMed.
- (a) S. J. Lee, S.-H. Cho, K. L. Mulfort, D. M. Tiede, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2008, 130, 16828 CrossRef CAS PubMed; (b) M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2013, 19, 10170 CrossRef CAS PubMed.
- (a) J. Yang, B. Chatelet, V. Dufaud, D. Hérault, S. Michaud-Chevallier, V. Robert, J.-P. Dutasta and A. Martinez, Angew. Chem., Int. Ed., 2018, 57, 14212 CrossRef CAS PubMed; (b) J. Yang, B. Chatelet, D. Hérault, J.-P. Dutasta and A. Martinez, Eur. J. Org. Chem., 2018, 5618 CrossRef CAS.
- C. Colomban, V. Martin-Diaconescu, T. Parella, S. Goeb, C. García-Simón, J. Lloret-Fillol, M. Costas and X. Ribas, Inorg. Chem., 2018, 57, 3529 CrossRef CAS PubMed.
- (a) O. Francesconi, A. Ienco, G. Moneti, C. Nativi and S. Roelens, Angew. Chem., Int. Ed., 2006, 45, 6693 CrossRef CAS PubMed; (b) B. Içli, N. Christinat, J. Tönnemann, C. Schüttler, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2009, 131, 3154 CrossRef PubMed.
- L. Cao, O. Caldararu, A. C. Rosenzweig and U. Ryde, Angew. Chem., Int. Ed., 2018, 57, 162 CrossRef CAS PubMed.
- J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901 CrossRef CAS PubMed.
- Z. Liu, Z. Shen, N. Zhang, W. Zhong and X. Liu, Catal. Lett., 2018, 148, 2709 CrossRef CAS.
- M. Otte, M. Lutz and R. J. M. Klein Gebbink, Eur. J. Org. Chem., 2017, 1657 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. CCDC 1868264 and 1876236. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc00437h |
| This journal is © The Royal Society of Chemistry 2019 |