
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cucurbituril-mediated quantum dot aggregates formed by aqueous self-assembly for sensing applications†
William J.
Peveler
 *ab,
Hui
Jia
c,
Tiffany
Jeen
b,
Kelly
Rees
b,
Thomas J.
Macdonald
d,
Zhicheng
Xia
b,
Weng-I Katherine
Chio
cde,
Suresh
Moorthy
c,
Ivan P.
Parkin
d,
Claire J.
Carmalt
d,
W. Russ
Algar
b and
Tung-Chun
Lee
*cd
*ab,
Hui
Jia
c,
Tiffany
Jeen
b,
Kelly
Rees
b,
Thomas J.
Macdonald
d,
Zhicheng
Xia
b,
Weng-I Katherine
Chio
cde,
Suresh
Moorthy
c,
Ivan P.
Parkin
d,
Claire J.
Carmalt
d,
W. Russ
Algar
b and
Tung-Chun
Lee
*cd
aDivision of Biomedical Engineering, School of Engineering, University of Glasgow, Glasgow, G12 8LT, UK. E-mail: william.peveler@glasgow.ac.uk
bDepartment of Chemistry, 2036 Main Mall, University of British Columbia, Vancouver, BC V6T 1Z1, Canada
cInstitute for Materials Discovery, University College London (UCL), UK. E-mail: tungchun.lee@ucl.ac.uk
dDepartment of Chemistry, University College London (UCL), 20 Gordon Street, London, WC1H 0AJ, UK
eSingapore Bioimaging Consortium (SBIC), Agency for Science Technology and Research (A*STAR), Singapore
First published on 12th April 2019
Abstract
Self-assembled nanoparticles have important applications in energy systems, optical devices and sensors, via the formation of aggregates with controlled interparticle spacing. Here we report aqueous self-assembly of rigid macrocycle cucurbit[7]uril (CB[7]) and fluorescent quantum dots (QDs), and demonstrate the potential of the system for efficient energy transfer and sensing of small molecules.
Controlled aggregation of nanoparticles into larger lattices can give rise to novel physiochemical properties that find numerous potential applications in energy systems and optical devices. Key to these applications is the control over interparticle spacing and particle composition. Controlled aggregation of semiconductor quantum dots (QDs) in particular was pioneered by Murray and Kagan, using controlled diffusion to achieve superlattice growth.1,2 This work led to a raft of studies to exploit the charge transport and corresponding optoelectronic properties of these materials in devices such as solar cells3,4 and optical sensors.5,6
Cucurbit[n]uril (CB[n]) is a rigid macrocycle of fixed height, 9 Å, and portal diameter varying from 2.4 to 6.9 Å as n varies from 5 to 8 glycoluril monomers. The CB portals are decorated with electron-rich carbonyl groups, and the cavity is highly hydrophobic.7 Previously, it has been demonstrated by the Scherman group that self-assembly of gold nanoparticles (Au NPs) can be mediated by supramolecular interactions between the CB portal and the Au NP surface.8–10 This binding interaction has been applied as molecular ‘glue’ in the controlled aggregation of Au NPs and Au nanorods to create plasmonic hotspots for surface-enhanced Raman spectroscopy (SERS).11–13
Similarly, small molecules,14 ligand–ligand interactions,15,16 and DNA17,18 have been used to mediate the controlled aggregation of QDs; however, the use of CB[n] as glue to modify the surface of nanomaterials beyond metal NPs19 and metal oxides20 remains less explored.
Herein, we report the first example of direct interaction between CB[7] and the surface of fluorescent metal chalcogenide (CdTe) QDs in aqueous solution. The binding interaction, together with the rigid molecular construct of CB[7], can guide the formation of precise photonic nanojunctions via a self-assembly process in water, enabling efficient energy transfer between QDs, and we characterise these structures with a range of spectroscopic techniques. The aggregation approach can be applied to create energy transfer cascades between QDs of different sizes. Moreover, CB[7] appears to retain some of its host–guest binding capability after associating with the QD surface, allowing surface enrichment of analyte molecules, including those which only interact weakly with QDs. We demonstrate a proof-of-concept sensing experiment, enhancing the sensitivity of fluorescent QDs towards nitroaromatics for “turn-off” detection.
Our initial experiments into QD aggregation with CB[n] utilised CB[7] (Fig. 1A) for its relatively high water solubility and larger cavity size (useful in later host–guest mediated sensing studies),7 and aqueous CdTe QDs for their bright emission and good colloidal-stability in the aqueous phase.21 The latter were synthesised in various sizes, and thus emission colours, with a 3-mercaptopropionic acid (MPA) capping layer. Red, orange, and green samples were produced as described in the ESI† and denoted as QDR, QDO and QDG, respectively.
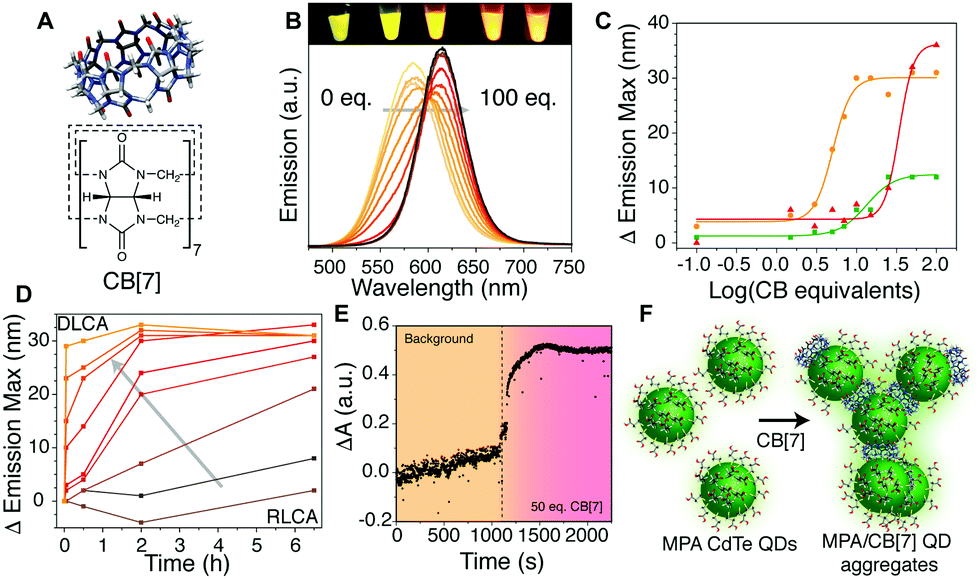 | ||
| Fig. 1 (A) Molecular structure of cucurbit[7]uril. (B) Brightening and red-shift of QDO with increasing CB[7] equivalents. Inset image shows aqueous QDO with (L to R) 1, 5, 10, 25 and 50 eq. CB[7] under UV illumination. (C) Change in emission maximum wavelength of QDR, QDO, and QDG (15 μM) with increasing CB[7] equivalents measured 5 min after addition. (D) Change in λem maximum for a sample of QDO over time with 0, 0.1, 1.5, 3, 5, 7, 10, 15 and 50 eq. CB[7] (following arrow) showing a change from RLCA to DLCA (see main text). (E) Aggregation measured by optical extinction increase on adding 50 eq. of CB[7] to QDR. (F) Schematic of ligand exchange by CB[7] on a QD surface causing aggregation and enabling energy transfer. | ||
A sample of single emission-colour QDs at fixed concentration was mixed with different concentrations of CB[7] in deionised water. Above a critical threshold of about 1 CB molecule per QD (Fig. 1B and C), a red-shift of 10–35 nm in emission wavelength was observed. This fluorescence evolution was attributed to formation of QD aggregates. Through energy transfer from slightly smaller QDs to the slightly larger QDs within the population, there is a red-shift and brightening of longer wavelengths, consistent with other reports on similar systems.14,22,23 The emission intensity was also modified with a decrease in intensity followed by an increase as aggregation proceeded (Fig. S1–S6, ESI† for experimental details).
The aggregates showed a tendency to grow over time (Fig. 1D), with a growth rate related to the equivalents of CB[7] added. At low equivalents (ca. 1–5 eq.) the growth of larger aggregates (corresponding to a larger red-shift in emission) took several hours to occur with kinetics resembling that of reaction limited colloidal aggregation (RLCA).9,24 In the case of a large excess of CB (>30 eq. per QD, ca. 15 μM QD concentration), aggregates formed rapidly through diffusion limited colloidal aggregation (DLCA) and started to flocculate out of solution within 12 h. To obtain a higher temporal resolution, the fast DLCA regime was also probed by measuring light absorption/scattering of samples upon addition of a large CB[7] excess (Fig. 1E). An immediate increase in optical extinction was observed upon CB[7] addition, and it then reduced slightly over time as the larger aggregates formed and precipitated. Dynamic light scattering measurements could be obtained for the larger aggregate sizes, showing small aggregates of 10–100 nm, growing to over 1000 nm with more CB equivalents (Fig. S7, ESI†). We postulate that the CB[7] is able to exchange with the MPA ligands on the surface of the QDs, leading to aggregation of the QDs via binding by one or both portals of the CB[7] (Fig. 1F), similar to the case of other nanoparticles.10,25 The ratio of QDs to CB[7] and total concentration will determine the extent and rate of aggregation occurring and limit the size of the clusters formed.
Estimation of CBs per unit surface area of QD reveals that, at CB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :QD ratios greater than 7:1–15:1, there were more CBs present than could fit on an ideally packed surface (full calculations given in Table S1, ESI†). IR spectra (Fig. S8, ESI†) showed residual MPA signals still visible in washed aggregates formed with 50 eq. of CB[7]. This suggests only a partial displacement of more strongly appended thiol ligands by more weakly binding CB[7]. If the CB per unit QD surface area is high enough (20–30 times molar excess, dependent on the size of the QD – Fig. 1C), aggregation is almost immediate. At ratios lower than a critical excess, aggregation is kinetically limited and the lower the equivalency, the slower the effect seen. The aggregates formed with lower equivalencies of CB tend to remain stable in aqueous solution for days or weeks, whilst those with higher equivalencies will continue to grow and precipitate. The inflection points observed in Fig. 1C vary due to both particle surface area, and also influence of sample heterogeneity and thiol coverage. A similar aggregation effect was observed with MPA-capped CdSe/ZnS QDs, but with less red-shifting, due to the much more homogenous NP population (Fig. S9, ESI†).
:QD ratios greater than 7:1–15:1, there were more CBs present than could fit on an ideally packed surface (full calculations given in Table S1, ESI†). IR spectra (Fig. S8, ESI†) showed residual MPA signals still visible in washed aggregates formed with 50 eq. of CB[7]. This suggests only a partial displacement of more strongly appended thiol ligands by more weakly binding CB[7]. If the CB per unit QD surface area is high enough (20–30 times molar excess, dependent on the size of the QD – Fig. 1C), aggregation is almost immediate. At ratios lower than a critical excess, aggregation is kinetically limited and the lower the equivalency, the slower the effect seen. The aggregates formed with lower equivalencies of CB tend to remain stable in aqueous solution for days or weeks, whilst those with higher equivalencies will continue to grow and precipitate. The inflection points observed in Fig. 1C vary due to both particle surface area, and also influence of sample heterogeneity and thiol coverage. A similar aggregation effect was observed with MPA-capped CdSe/ZnS QDs, but with less red-shifting, due to the much more homogenous NP population (Fig. S9, ESI†).
To further investigate the QD–CB[7] interaction, NMR studies were performed. 1H NMR data from QD solutions in D2O with increasing CB[7] concentration showed several MPA molecular environments between 2.3 and 3.0 ppm (arising from the CH2–CH2 region – Fig. 2A and Fig. S10, ESI†). We employed 2D COSY experiments to identify two pairs of signals 2.45/2.75 ppm and 2.35/2.57 (Fig. S11, ESI†). We identified that the larger sharp triplet pair (2.45/2.75 ppm) likely originates from the presence of oxidised MPA dimer in solution, using further 13C NMR spectroscopy and mass spectrometry (Fig. S12 and S13, ESI†).26 The second sharp pair observed (2.35/2.57 ppm) appeared to overlay with the free MPA signals measured at the same pH, but with a considerably broader linewidth, which suggests transient binding of free MPA on the QD surfaces. We also observe a third pair (ca. 2.6 and 2.9 ppm, Fig. 2A), which is considerably broadened and featureless, and therefore does not show COSY connectivity in the 2D analysis. This pair of signals is shifted downfield in comparison to the “free” MPA signals and is hypothesised to originate from the MPA molecules tightly bound to the QD surfaces through Cd–S bonds. The extreme broadness of this pair arises from both the slow motion of the sizeable QD–MPA conjugates and the multiple possible binding environments for MPA on the QD surfaces.
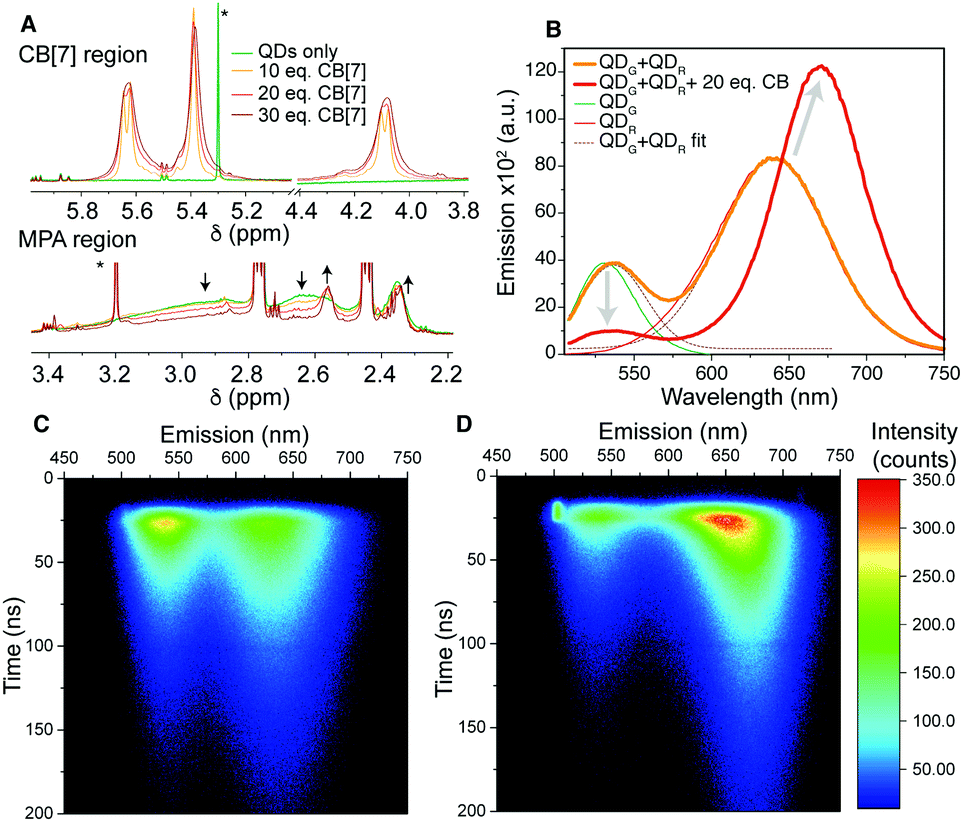 | ||
| Fig. 2 (A) 1H NMR regions of 15 μM QDR in D2O showing the MPA broad signal loss and changes to the CB signals. Arrows indicate change with increasing CB[7] concentration and * indicates solvent residues (full spectra in Fig. S10, ESI†). Energy transfer study: (B) emission spectra for a QDG and QDR mixture with and without 20 eq. of CB[7], showing the red-shift and loss of the green QD signal. (C) 2D maps of emission wavelength and emission decay for un-aggregated and (D) aggregated QDs. The bright region at 20 ns/500 nm is scattered excitation light. | ||
Once CB[7] is added to the QD solution, we observe three further signals downfield (4.10, 5.40 and 5.65 ppm, Fig. 2A). These signals are considerably broadened in comparison to free CB[7] signals, which suggests interaction with the QD surface. The CB signals broaden further on increasing CB concentration and we consider the increased concentration to drive more CB on to the QD surface, and the size of the QD aggregates themselves increases, slowing motion on the NMR timescale and trapping more CB. Saturation Transfer Difference (STD) NMR spectroscopy was utilised to further evidence the binding of CB[7] to QD surfaces (Fig. S14, ESI†).27,28
To investigate steady-state energy transfer within the CB–QD aggregates, we mixed two differently sized QDs with green and red emissions with spectral overlap between the emission of the green QD and the absorption of the red QD. On mixing in solution, there was no change in spectral output. Both QD emissions could be deconvoluted and had similar emission peak positions and intensities to before mixing (Fig. 2B). However, upon addition of 20 eq. of CB[7], a red-shift of the peaks was observed and, significantly, there was quenching of the green QD emission and brightening of the red QD emission. This result illustrated that intimate mixing and ‘gluing’ of the different QDs enabled efficient energy transfer. A more complex three emission cascade was also achieved (Fig. S15, ESI†).
Energy transfer was used to probe the dynamic nature of the aggregates. A mixture of two pre-formed QD aggregates of different colours displayed energy transfer over time, indicating exchange between aggregates occurred, with incorporation of both colours of QDs into the final aggregates (Fig. S16, ESI†).
To further understand the energy transfer in the CB–QD aggregates, time-resolved emission of a QDG/QDR mixture was studied with and without CB[7] (Fig. 2C and D). Emission lifetimes were calculated for the spectral range of the entire QDG emission peak (510–570 nm, τav = 37 ns) and for three regions of the broader QDR peak (blue-edge, 600–630 nm, 47 ns; centre, 630–670 nm, 52 ns; red-edge, 670–730 nm, 57 ns) (Fig. S17, S18 and Table S2, ESI†).29 After aggregation induced by CB[7], a third, short component (2.6 ns) appeared in the QDG lifetime (when fixing the original two components), and the average lifetime of the blue-edge of the QDR decay decreased (35 ns). The central spectral region for the QDR became multi-exponential due to convolution of the green-to-red and red-to-red energy transfer processes, and was fitted with a shortened component (30 ns) and lengthened component (95 ns). The red-edge of the QDR decay remained approximately constant in lifetime, consistent with this sub-population of largest QDs acting as the final acceptor of transferred energy. It was noticeable that for this red-edge emission the rise time increased, consistent with increased sensitisation of emission from energy transfer rather than direct excitation by the laser pulse.
Having investigated the aggregation effect, we sought to take advantage of the host–guest chemistry available to CB[7] and use this system for fluorometric sensing. We hypothesised that if an electron withdrawing molecule could be encapsulated by the CB cavity, then the system's fluorescence would be perturbed and act as a reporter of binding.30 As a proof-of-concept for solution sensing, we targeted nitrated aromatics, an important class of volatile organic compound that are often found in explosive formulations. Nitrobenzene (NB) and 2,4-dinitrotoluene (DNT) were mixed with aqueous solutions of CB[7] and the QDs. The QD aggregates formed in the presence of analyte, evidenced by the normal red-shifting of the emission band (Fig. 3A and Fig. S19, ESI†). For DNT addition, ca. 4 times more quenching of the QD occurred in the presence of CB[7] than without (Fig. S20, ESI†). Reduced red-shift of the QD emission at high analyte concentrations was observed, hinting that full aggregation may be hindered. This may be due to the analyte blocking one of the two CB portals, preventing complete aggregation and was particularly marked at the highest concentrations tested (0.35 and 0.7 mM) where there was calculated to be >1 DNT per CB[7] in solution (Tables S3 and S4, ESI†). The prevention of aggregation was further evidenced by the mixture of CB[7] with red and green QDs in the presence of 0.35 mM DNT, showing a distinct increase in green emission in the presence of the analyte as well as the expected fluorescence shifting and quenching (Fig. 3B). A cartoon of the sensing process is given in Fig. 3C and expanded on in Fig. S21 (ESI†).
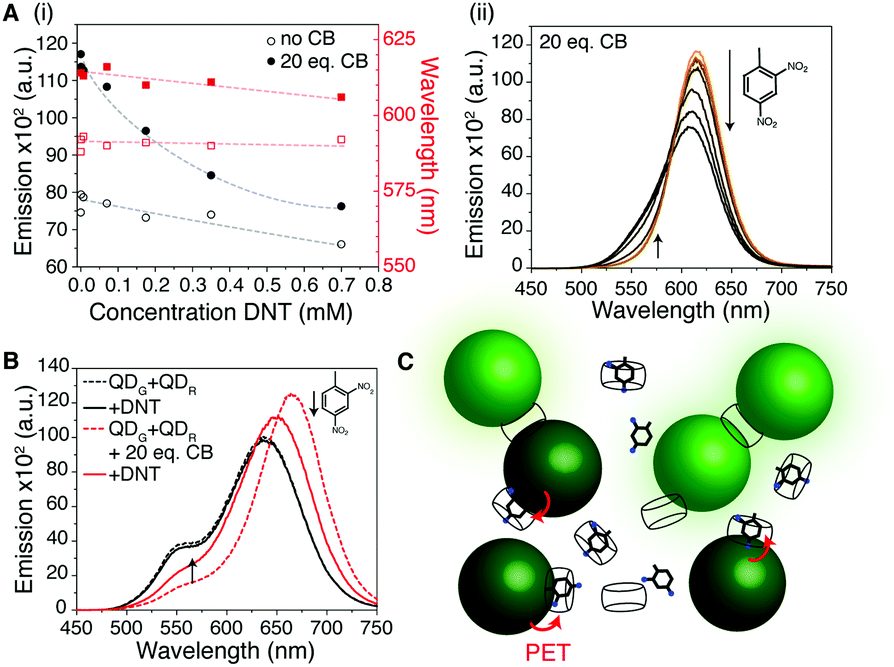 | ||
| Fig. 3 (A) DNT (saturated aqueous solution) added into the mixture of CB[7] and QDO exhibits enhanced quenching over that observed in the absence of CB[7]. Black circles correspond to Δintensity and red squares Δλem max. Dotted lines are to guide the eye. (B) In a two-QD system with 0.35 mM DNT, increased quenching of the QDR peak is observed in the presence of CB[7] but an increase in PL is observed for the QDG emission. (C) Cartoon of the sensing process. Photoinduced electron transfer (red arrows) from the QD to the electron-withdrawing DNT, held in close proximity by CB[7], causes efficient quenching. MPA not shown for clarity. | ||
The importance of the electron-withdrawing nature of the analyte was confirmed by a lack of any quenching using toluene in a control experiment (Fig. S22, ESI†). Whilst it is possible that some of the quenching we observe is due to non-specific trapping of the electron withdrawing analyte in the CB aggregates, rather than host–guest binding, simulations of DNT–CB interactions suggest such binding is possible. The short distances that photoinduced electron transfer (PET – the likely quenching mechanism here) operates on, suggest tight binding on the surface of the QD via the CB is plausible.
In conclusion, we have provided evidence that CB[7] mediates aggregation of QDs in aqueous media through portal binding. The aggregate size changes in response to the ratio of CB to QD, and thus we suggest, the extent of CB displacement of MPA. NMR, IR and emission spectroscopy provide evidence for the hypothesised CB-induced aggregation mechanism. We determined that aggregates formed from 10–20 equivalents of CB per QD were colloidally stable enough for further solution studies. Energy transfer cascades occur within the aggregates with fast (kET ≈ 3.6 × 108 s−1) energy transfer from smaller to larger QDs. We propose that QD-bound CB[7] remains supramolecularly active, and can enhance the quenching of QDs several-fold via a surface enrichment mechanism. These findings provide a proof-of-concept for new possibilities to exploit host–guest chemistry in aqueous, soft nanophotonic systems, and for self-assembling precise yet dynamic photonic nanostructures for selective sensors and energy-harvesting devices.
WJP thanks the Izaak Walton Killam Memorial Fund for a Fellowship and the University of Glasgow for an LKAS Fellowship. TJM thanks the Ramsay Memorial Trust. SM and TCL are grateful for a Leverhulme Trust Research Project Grant (RPG-2016-393). WIKC and TCL are grateful for a Studentship from the A*STAR-UCL Research Attachment Programme via the EPSRC M3S CDT (EP/L015862/1). WRA, TJ, KR and WJP thank the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Foundation for Innovation, and the University of British Columbia for support of their research. TJ is grateful for a NSERC CREATE NanoMat fellowship. KR is grateful for a UBC four-year fellowship. WRA gratefully acknowledges a Canada Research Chair (Tier 2), a Michael Smith Foundation for Health Research Scholar Award, and an Alfred P. Sloan Fellowship. The authors thank Dr Saeid Kamal for assistance with measurements in the Laboratory for Advanced Spectroscopy and Imaging Research at UBC.
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Science, 1995, 270, 1335–1338 CrossRef CAS.
- E. V. Shevchenko, D. V. Talapin, N. A. Kotov, S. O'Brien and C. B. Murray, Nature, 2006, 439, 55–59 CrossRef CAS PubMed.
- C. R. Kagan, E. Lifshitz, E. H. Sargent and D. V. Talapin, Science, 2016, 353, aac5523 CrossRef PubMed.
- I. E. Rauda, L. C. Saldarriaga Lopez, B. A. Helms, L. T. Schelhas, D. Membreno, D. J. Milliron and S. H. Tolbert, Adv. Mater., 2013, 25, 1315–1322 CrossRef CAS.
- K. Chou and A. Dennis, Sensors, 2015, 15, 13288–13325 CrossRef PubMed.
- X. Shi, S. Dong, M. Li, X. Liu, Q. Zhang, W. Zhao, C. Zong, Y. Zhang and H. Gai, Chem. Commun., 2015, 51, 2353–2356 RSC.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- T.-C. Lee and O. A. Scherman, Chem. – Eur. J., 2012, 18, 1628–1633 CrossRef CAS PubMed.
- R. W. Taylor, T.-C. Lee, O. A. Scherman, R. Esteban, J. Aizpurua, F. M. Huang, J. J. Baumberg and S. Mahajan, ACS Nano, 2011, 5, 3878–3887 CrossRef CAS PubMed.
- T.-C. Lee and O. A. Scherman, Chem. Commun., 2010, 46, 2438–2440 RSC.
- S. T. Jones, R. W. Taylor, R. Esteban, E. K. Abo-Hamed, P. H. H. Bomans, N. A. J. M. Sommerdijk, J. Aizpurua, J. J. Baumberg and O. A. Scherman, Small, 2014, 10, 4298–4303 CAS.
- S. Mahajan, T.-C. Lee, F. Biedermann, J. T. Hugall, J. J. Baumberg and O. A. Scherman, Phys. Chem. Chem. Phys., 2010, 12, 10429–10433 RSC.
- S. Kasera, L. O. Herrmann, J. D. Barrio, J. J. Baumberg and O. A. Scherman, Sci. Rep., 2014, 4, 6785 CrossRef CAS PubMed.
- E. Cohen, I. Gdor, E. Romero, S. Yochelis, R. van Grondelle and Y. Paltiel, J. Phys. Chem. Lett., 2017, 8, 1014–1018 CrossRef CAS PubMed.
- M. Wu, P. Mukherjee, D. N. Lamont and D. H. Waldeck, J. Phys. Chem. C, 2010, 114, 5751–5759 CrossRef CAS.
- J. Liu, X. Yang, K. Wang, X. He, Q. Wang, J. Huang and Y. Liu, ACS Nano, 2012, 6, 4973–4983 CrossRef CAS PubMed.
- S. J. Barrow, X. Wei, J. S. Baldauf, A. M. Funston and P. Mulvaney, Nat. Commun., 2012, 3, 1275 CrossRef PubMed.
- L. Zhang, S. R. Jean, S. Ahmed, P. M. Aldridge, X. Li, F. Fan, E. H. Sargent and S. O. Kelley, Nat. Commun., 2017, 8, 381 CrossRef PubMed.
- X. Lu and E. Masson, Langmuir, 2011, 27, 3051–3058 CrossRef CAS PubMed.
- F. Benyettou, K. N. Nono, M. Jouiad, Y. Lalatonne, I. Milosevic, L. Motte, J. C. Olsen, N. Saleh and A. Trabolsi, Chem. – Eur. J., 2015, 21, 4607–4613 CrossRef CAS PubMed.
- D. P. Tran, T. J. Macdonald, B. Wolfrum, R. Stockmann, T. Nann, A. Offenhäusser and B. Thierry, Appl. Phys. Lett., 2014, 105, 231116 CrossRef.
- S. A. Crooker, J. A. Hollingsworth, S. Tretiak and V. I. Klimov, Phys. Rev. Lett., 2002, 89, 186802 CrossRef CAS PubMed.
- C. R. Kagan, C. B. Murray, M. Nirmal and M. G. Bawendi, Phys. Rev. Lett., 1996, 76, 1517–1520 CrossRef CAS PubMed.
- M. Y. Lin, H. M. Lindsay, D. A. Weitz, R. C. Ball, R. Klein and P. Meakin, Nature, 1989, 339, 360–362 CrossRef CAS.
- Y. Sun, W. Zhang, B. Wang, X. Xu, J. Chou, O. Shimoni, A. T. Ung and D. Jin, Chem. Commun., 2018, 54, 3851–3854 RSC.
- X. B. Li, Z. J. Li, Y. J. Gao, Q. Y. Meng, S. Yu, R. G. Weiss, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2014, 53, 2085–2089 CrossRef CAS PubMed.
- M. Mayer and B. Meyer, Angew. Chem., Int. Ed., 1999, 38, 1784–1788 CrossRef CAS PubMed.
- Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211–1221 CrossRef CAS.
- A. L. Rogach, T. Franzl, T. A. Klar, J. Feldmann, N. Gaponik, V. Lesnyak, A. Shavel, A. Eychmüller, Y. P. Rakovich and J. F. Donegan, J. Phys. Chem. C, 2007, 111, 14628–14637 CrossRef CAS.
- W. J. Peveler, A. Roldan, N. Hollingsworth, M. J. Porter and I. P. Parkin, ACS Nano, 2016, 10, 1139–1146 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional methods and characterisation. See DOI: 10.1039/c9cc00410f |
| This journal is © The Royal Society of Chemistry 2019 |