
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A gene-targeted polymerase-mediated strategy to identify O6-methylguanine damage†
Claudia M. N.
Aloisi
 ,
Shana J.
Sturla
and
Hailey L.
Gahlon
*
,
Shana J.
Sturla
and
Hailey L.
Gahlon
*
Department of Health Sciences and Technology, ETH Zürich, Schmelzbergstrasse 9, 8092 Zürich, Switzerland. E-mail: hailey.gahlon@hest.ethz.ch
First published on 6th March 2019
Abstract
Detecting DNA adducts in cancer genes is important for understanding cancer etiology. This study reports a strategy to identify the mutagenic DNA adduct O6-methylguanine in K-Ras. The strategy involves selective replication past a synthetic primer when placed opposite O6-methylguanine. Future work can apply this approach to other cancer-relevant genes.
DNA adducts contribute to cancer initiation and can be biomarkers of carcinogenesis as well as cancer therapy efficacy. Strategies to detect DNA damage often describe global damage levels, e.g. mass spectrometry, immunological assays and 32P-postlabeling, and information regarding their genomic sequence context is missing.1–5 In recent years, various methods have been developed to report on damage in a sequence-specific context, for instance single-molecule approaches like nanopore and SMRT sequencing as well as whole genome sequencing.6–14 Recently, the emergence of epigenetic factors being important in driving biological responses to chemicals15,16 emphasizes the need for methods to define the presence of DNA damage in particular genes. However, DNA damage sequencing approaches are only recently emerging and have not been generalized to many forms of DNA damage. O6-Methylguanine (O6-MeG) is a mutagenic DNA adduct that is formed upon exposure to endogenous and exogenous alkylating agents, e.g. N-nitroso-N-methylurea and N-nitrosodimethylamine.17,18 The K-Ras gene is mutated in 43% of colon cancer cases,19 for which nitrosamine exposure may be a contributing factor, but lack of strategies to investigate the formation of O6-MeG in K-Ras and mutation hotspot loci limits the capacity to relate mutagenesis with specific DNA methylation events. This work describes a proof of principle approach to detect the mutagenic adduct O6-MeG in the K-Ras gene sequence.
In previous studies addressing structural requirements for extension from a modified primer terminus, we reported that Dpo4, a Y-family DNA polymerase, elongated past O6-alkylguanine DNA adducts when paired opposite a synthetic nucleoside analogue.20,21 This process was effective for elongation past O6-MeG when paired opposite the hydrophobic nucleoside analogue Benzi, designed to pair preferentially opposite O6-alkylguanines.20–23 When Benzi was paired opposite G, however, Dpo4 stalled and the primer was not elongated.21 We envisioned that the selective extension past O6-MeG as compared to G could be the basis of a strategy to detect O6-MeG. However, preliminary studies with oligonucleotides in the K-Ras sequence (Table 1) revealed that Dpo4 elongated past Benzi paired opposite both O6-MeG and G similarly (Fig. S4, ESI†). On the other hand, replacing Dpo4 with either KlenTaq or a previously reported mutant of KlenTaq M474K showed selective extension, i.e. Benzi primer was only enzymatically elongated when paired opposite O6-MeG and not G (Fig. 1 and Fig. S6, ESI†). In this study, we have developed a strategy to detect O6-MeG in the target gene sequence of K-Ras by using a 3′-Benzi-modified primer that hybridizes opposite damaged and undamaged templates and an engineered DNA polymerase that is selective and efficient for extension of modified primers (Scheme 1).
| Name | DNA sequence (5′–3′)a |
|---|---|
| a G* = O6-MeG, underlined bases = codon 12 of K-Ras, P = Benzi, N = (T)21 that is represented as a 5′-overhang (green line) in Scheme 1A. | |
| G template | GTA GTT GGA GCT ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) ![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) GGC GTA GGC AAG GGC GTA GGC AAG |
| O 6-MeG template | GTA GTT GGA GCT * GGC GTA GGC AAG |
| Benzi primer 1 | CTT GCC TAC GCC AP |
| Benzi primer 2 | NCTT GCC TAC GCC AP |
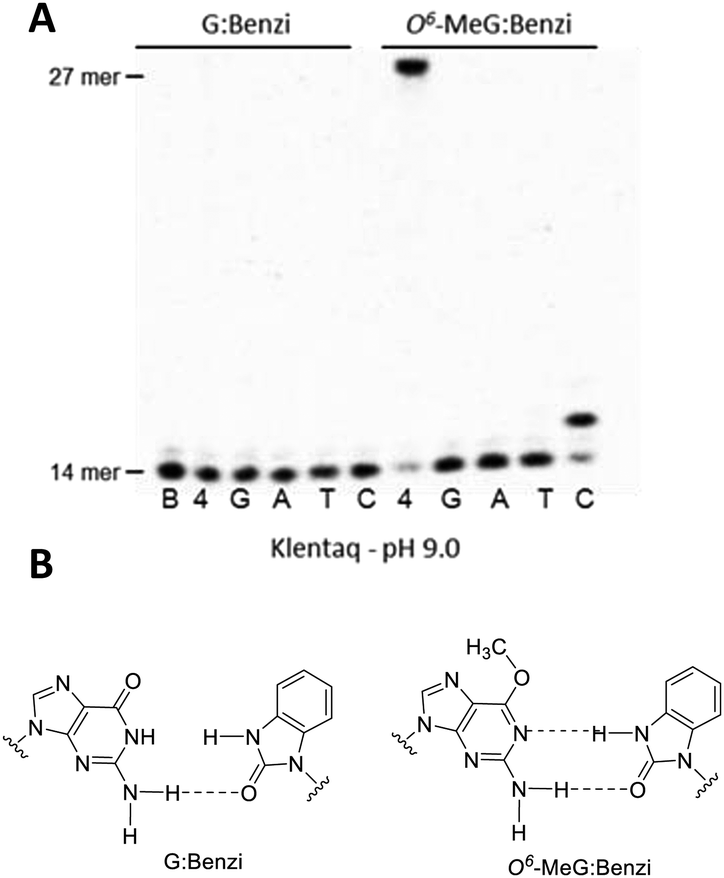 | ||
| Fig. 1 (A) KlenTaq-mediated elongation past G:Benzi (left) and O6-MeG:Benzi (right) at 55 °C for 10 min at pH 9.0 (lanes in the gel are designated as follows: B = 14 mer blank, 4 = all 4 canonical dNTPs, and the single nucleotide conditions include: G = only dGTP, A = only dATP, T = only dTTP, and C = only dCTP). (B) Proposed base pairing interactions for Benzi opposite G and O6-MeG. | ||
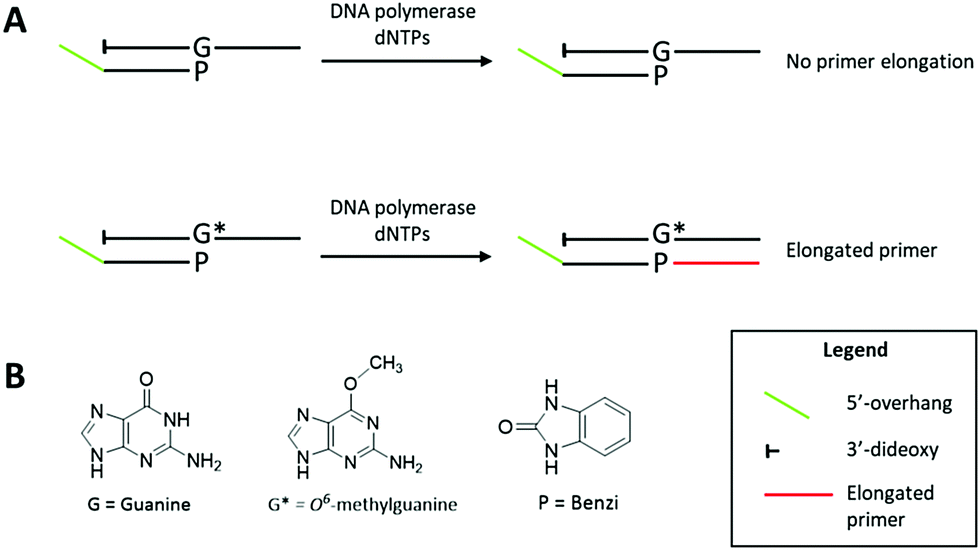 | ||
| Scheme 1 (A) KlenTaq elongates specifically past O6-MeG:Benzi termini and does not elongate past G:Benzi in the K-Ras gene sequence. This discrimination provides a mean to detect O6-MeG by linear DNA amplification. Extension products are separated by polyacrylamide gel electrophoresis and visualized by SYBR Gold staining. The elongated primer (red region) is distinguishable from the O6-MeG- and G-containing templates by the presence of a 5′ overhang (green region) on the primer. The template contains a 3′-dideoxy modification to prevent undesired extension of the template. (B) Chemical structures of guanine (G), O6-MeG (G*) and Benzi (P). | ||
The potential of selective enzymatic extension past O6-MeG in a target gene was tested in the sequence of codon 12 of the K-Ras gene, a mutational hotspot locus that is frequently mutated in colorectal cancers.19 We performed primer extension experiments with 3′-Benzi-modified primers and templates with the sequence of K-Ras gene where the second base of codon 12 contains a G or O6-MeG (Table 1). Dpo4 elongated past Benzi paired opposite both O6-MeG and G similarly, suggesting a gap in the potential to extend the strategy to other gene-related sequences (Fig. S4, ESI†). We tested the influence of the enzyme on this process, replacing Dpo4 with bypass-proficient engineered enzymes, KlenTaq and its M747K mutant.23,24 The change in the enzyme resulted in selective extension, whereby Benzi primer was only enzymatically elongated when paired opposite O6-MeG but not G (Fig. 1 and Fig. S6, ESI†).
The ability for extension by KlenTaq past Benzi-containing primers paired opposite G and O6-MeG was evaluated by further primer extension analysis. KlenTaq elongated selectively past Benzi paired opposite O6-MeG, and not G, in the K-Ras DNA sequence (Table 1). For these experiments, KlenTaq DNA polymerase (25 units), DNA (10 nM) and dNTPs (10 μM) were incubated at 55 °C for 10 min. For O6-MeG:Benzi DNA, KlenTaq extended in an error-free manner and only dCTP was inserted under single nucleotide conditions (Fig. 1). In the case of all four dNTPs, KlenTaq performed full-length extension for O6-MeG:Benzi and no extension was observed for G:Benzi. KlenTaq efficiently elongated past O6-MeG:Benzi DNA in the presence of all four dNTPs (90% elongation, Fig. 1A). Analogous primer extension assays were performed with a lower concentration of KlenTaq (2.5 units) and less elongation was observed for the O6-MeG:Benzi DNA (∼65% extension, Fig. S5, ESI†).
Elongation past Benzi-containing DNA was also tested with the KlenTaq mutant M747K, reported to have an increased proclivity for lesion bypass. This engineered polymerase is thought to increase favourable electrostatic interactions between the DNA backbone and the enzyme, thus promoting damage bypass.25,26 For the primer extension reactions, KlenTaq M747K (20 nM), DNA (10 nM) and dNTPs (10 μM) were incubated at 55 °C for 10 min (Fig. S6, ESI†). For G:Benzi DNA, KlenTaq M747K did not catalyze DNA synthesis (Fig. S6, ESI†). Conversely, for O6-MeG:Benzi DNA, KlenTaq M747K elongated by correctly inserting dCMP (Fig. S6, ESI†). Also, for O6-MeG:Benzi, KlenTaq M747K elongated the primer to the full length of the template in the presence of all 4 dNTPs (∼100%, Fig. S6, ESI†). KlenTaq M747K-mediated elongation was also tested at 72 °C, however, no full-length extension products were observed for either G:Benzi or O6-MeG:Benzi (Fig. S7, ESI†). A small percentage of dCMP (∼10%) was inserted for O6-MeG:Benzi (Fig. S7, ESI†). It is likely that at 72 °C the DNA is not hybridized, as the theoretical Tm is ∼50 °C. Therefore, temperatures below 72 °C are necessary for optimal DNA polymerase-mediated elongation. A slight reduction in selectivity past O6-MeG:Benzi vs. G:Benzi was observed for KlenTaq M747K (Fig. S6, ESI†) compared to wild type KlenTaq (Fig. 1). Therefore, wild type KlenTaq was used in further experiments.
Having established wild type KlenTaq as the preferred enzyme for selective extension from DNA templates containing O6-MeG, we tested if the Benzi primer could be linearly amplified in the presence of G and O6-MeG templates. For these studies, a modified Benzi oligonucleotide (Benzi primer 2, Table 1) was synthesized (Fig. S3, ESI†) with a 21 nt 5′-overhang to enable resolution of amplicons (48 nt extended primer) from the initial template (27 nt). To ensure that the observed 48 nt oligo signal was exclusively a result of elongation of the Benzi primer, templates with the 3′-dideoxy ends were used. To achieve the highest selectivity and enzyme efficiency, we optimized temperature and time of each reaction step, cycle number, concentration of primer and templates. In addition, we evaluated the effect of varying concentrations of Mg2+ ions (Fig. S8, ESI†). Final conditions for the amplification reactions consisted of Benzi primer 2 (200 nM) incubated with either O6-MeG or G template (2 nM each template), KlenTaq (20 nM) and all four dNTPs (250 μM). Alternating cycles (35) of DNA melting (95 °C, 30 s), annealing (40 °C, 30 s) and extension step (55 °C, 30 s) were performed and products were separated on polyacrylamide gels and visualized by staining with SYBR gold. Under these linear amplification conditions, selective elongation for O6-MeG:Benzi DNA was observed (Fig. 2A), corroborating findings from the primer extension data (Fig. 1).
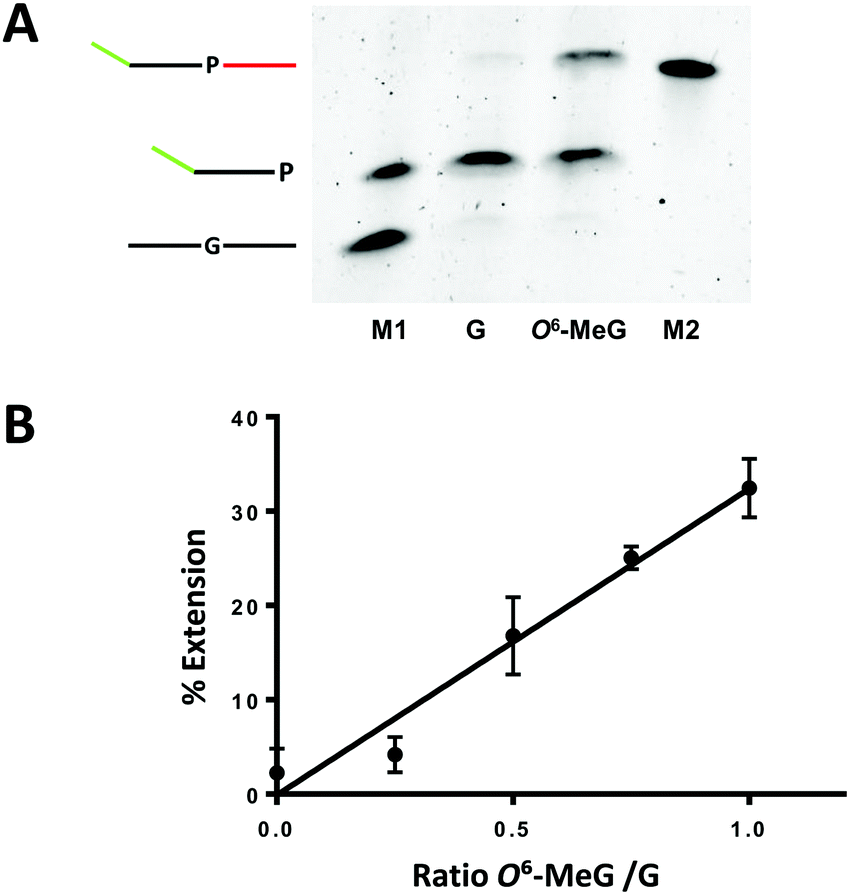 | ||
| Fig. 2 KlenTaq-mediated elongation past G:Benzi and O6-MeG:Benzi with Benzi primer 2 (Table 1). Linear amplification conditions comprised of (1) denaturation at 95 °C for 2 min; (2) 35 cycles alternating steps of denaturation at 95 °C for 30 s, annealing at 40 °C for 30 s, extension at 55 °C for 30 s; (3) final extension at 55 °C for 3 min; pH 9, 1XKTQ buffer. (A) Gel separated amplicons (M1 = marker lane with 27 mer G template and 35 mer Benzi primer 2; G = reaction with G template; O6-MeG = reaction with O6-MeG template; M2 = marker lane with 48 mer extended primer). Note: initial templates are not detected by SYBR Gold staining as they are below the limit of detection. (B) Linear amplification was performed in the presence of G and O6-MeG templates with increasing concentrations of O6-MeG template and a constant concentration of G template (2 nM). All reactions were performed in three independent technical replicates (y = 32.49x −0.11; R2 = 0.97). | ||
To test if the selective elongation from O6-MeG:Benzi could be a viable strategy for identifying O6-MeG, we performed linear amplification studies in a mixture of damaged and undamaged DNA. Here, the concentration of G template was constant (2 nM) while increasing the concentrations of O6-MeG (0, 0.5, 1, 1.5, 2 nM). In the presence of a mixture of DNA templates, the signal increased as a function of increasing concentrations of O6-MeG (Fig. 2B). The ratio of O6-MeG to G plotted against the corresponding amount of extension showed a linear relationship (Fig. 2B). In a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture of O6-MeG:G, 15% of extension was observed and extension doubled (30%) with a 2-fold increase in the ratio of O6-MeG:G. These results demonstrate Benzi as an effective chemical marker to detect O6-MeG in a mixture of undamaged and damaged DNA.
:50 mixture of O6-MeG:G, 15% of extension was observed and extension doubled (30%) with a 2-fold increase in the ratio of O6-MeG:G. These results demonstrate Benzi as an effective chemical marker to detect O6-MeG in a mixture of undamaged and damaged DNA.
To understand structural interactions that contribute to the preferred extension of Benzi paired opposite O6-MeG by KlenTaq, we performed computational modelling studies. Previous X-ray data with Benzi as an incoming triphosphate (BenziTP) paired opposite O6-MeG (i.e. ternary complex; Fig. 3A) showed BenziTP pairs opposite O6-MeG with two hydrogen bond interactions.26 Starting from the ternary complex crystal structure (PDB ID: 3RTV)26 with wild type KlenTaq, undamaged primer:template DNA, and an incoming dCTP we (1) removed the incoming dCTP (2) replaced the terminal 3′ nucleotide on the primer strand with Benzi, and (3) inserted G and O6-MeG in the template (Fig. 3B) to reflect the configuration of the experiments described above.
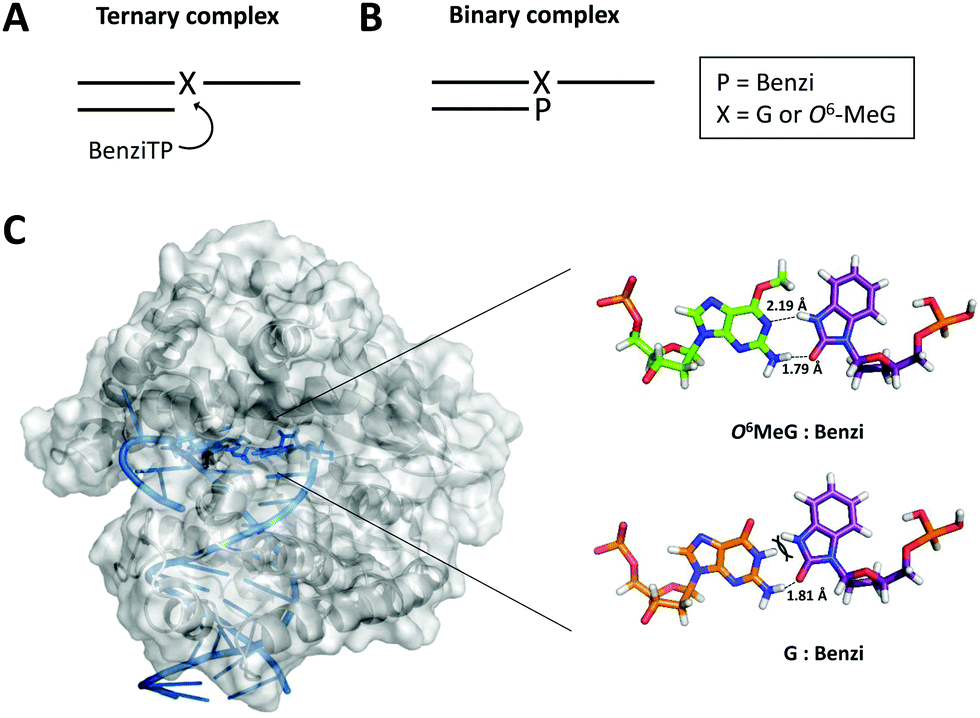 | ||
| Fig. 3 (A) Scheme of the ternary complex previously crystallized for X-ray analysis (X = G or O6-MeG). (B) Scheme of the binary complex with Benzi covalently bound to the 3′ end of the primer (P = Benzi). (C) KlenTaq (PDB ID: 3RTV) was energy minimized with Molecular Operating Environment software. The active site of KlenTaq (zoomed-in region) shows Benzi paired opposite O6-MeG (top) and G (bottom). Two hydrogen bond interactions were computed for O6-MeG:Benzi and one hydrogen bond for G:Benzi. | ||
We evaluated the distance and orientation of the terminal base pairs in the active site of KlenTaq. First, we compared deoxyribose distances for O6-MeG:Benzi and the natural G:C base pairs. We found that the C1′–C1′ distance for the G:C base pair (10.5 Å) is comparable to the O6-MeG:Benzi base pair (10.4 Å and 10.2 Å for the methyl group in a distal and proximal conformation, respectively, Fig. S9, ESI†). These distances are similar to what was reported for the X-ray structure; O6-MeG:Benzi with a slightly longer C1′–C1′ distance of 11.0 Å and G:C of 10.6 Å.26 We speculate that the closer distance of O6-MeG:Benzi in our modelled data may result from the slight torsion that Benzi imposes when covalently bound to the DNA primer (binary complex, Fig. 3B). Next, we compared the orientation of Benzi in the enzymatic pocket when paired opposite O6-MeG and G. We observed minor rotations on amino acid residues; however, the overall orientation of Benzi was not sufficiently different when paired opposite damaged and undamaged DNA.
Overall energies and hydrogen bond interactions were determined for terminal base pairs in the active site of KlenTaq. We determined the computed energies of the terminal O6-MeG:Benzi and G:Benzi base pairs. These calculated energies take into account both van der Waals and hydrogen bond interactions. Computed energies of −0.3 kcal mol−1 for O6-MeG:Benzi and +26 kcal mol−1 for G:Benzi were determined. Further, we found that the hydrogen bond pattern in our model corroborates the previous X-ray data.26 For example, Benzi and O6-MeG interact by two hydrogen bonds; one hydrogen bond between the N1 on O6-MeG and the –NH donor on Benzi (2.19 Å, Fig. 3C) and another between the NH2 donor on O6-MeG and the carbonyl group on Benzi (1.79 Å when the methyl group is in the distal conformation, Fig. 3C; 1.96 Å in the proximal conformation, Fig. S9, ESI†). Further, G:Benzi displays a potential steric clash between the –NH moiety on Benzi and the –NH at the N1 position on G (Fig. 3C), possibly impeding KlenTaq catalysis. On the contrary, O6-MeG:Benzi is more analogous to the natural G:C Watson–Crick base pair, but with two hydrogen bonds. This more favourable configuration of O6-MeG:Benzi vs. G:Benzi may explain the observed preferential elongation by KlenTaq.
In conclusion, this work presents a first enzyme-based strategy reporting on the presence of O6-MeG in the cancer-relevant gene sequence of K-Ras by means of synthetic DNA-based amplification. This strategy works by identifying O6-MeG in a sequence-specific manner and, therefore, could be useful for linking modifications in particular genes with specific mutational outcomes, which is not possible using typical DNA adduct detection methods that generally lose information about adduct sequence. In this strategy, damage is detected in a pre-defined locus. Compared to genome-wide damage mapping strategies, this approach provides a targeted strategy that could be adapted to other cancer genes, thereby providing defined chemical resolution of damage in hotspot regions of genes. Future work will require assessing adduct selectivity, as well as addressing the need for increased sensitivity to detect damage at biologically relevant levels, which would require a companion enrichment strategy to assess low abundance forms of damage. Furthermore, this approach could be expanded to detect O6-MeG in other cancer-relevant gene sequences, thereby leading to a better understanding of the relationship between DNA damage and disease initiation and progression.
We acknowledge the Swiss National Science Foundation (31003A_156280), the European Research Council (260341) and the ETH Career Seed Grant (SEED-16 18-1).
Conflicts of interest
There are no conflicts to declare.References
- J. Guo, P. W. Villalta and R. J. Turesky, Anal. Chem., 2017, 89, 11728–11736 CrossRef CAS PubMed.
- R. Singh and P. B. Farmer, Carcinogenesis, 2006, 27, 178–196 CrossRef CAS PubMed.
- K. Brown, Methods Mol. Biol., 2012, 817, 207–230 CrossRef CAS PubMed.
- N. J. Jones, Methods Mol. Biol., 2012, 817, 183–206 CrossRef CAS PubMed.
- D. H. Phillips, P. B. Farmer, F. A. Beland, R. G. Nath, M. C. Poirier, M. V. Reddy and K. W. Turteltaub, Environ. Mol. Mutagen., 2000, 35, 222–233 CrossRef CAS PubMed.
- J. Hu, S. Adar, C. P. Selby, J. D. Lieb and A. Sancar, Genes Dev., 2015, 29, 948–960 CrossRef CAS PubMed.
- W. Li, J. Hu, O. Adebali, S. Adar, Y. Yang, Y.-Y. Chiou and A. Sancar, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6752–6757 CAS.
- J. Wu, M. McKeague and S. J. Sturla, J. Am. Chem. Soc., 2018, 140, 9783–9787 CrossRef CAS PubMed.
- J. Ding, M. S. Taylor, A. P. Jackson and M. A. M. Reijns, Nat. Protoc., 2015, 10, 1433 CrossRef CAS PubMed.
- J. Hu, J. D. Lieb, A. Sancar and S. Adar, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11507 CrossRef CAS PubMed.
- N. An, A. M. Fleming, H. S. White and C. J. Burrows, ACS Nano, 2015, 9, 4296–4307 CrossRef CAS PubMed.
- I. A. Trantakis, A. Nilforoushan, H. A. Dahlmann, C. K. Stäuble and S. J. Sturla, J. Am. Chem. Soc., 2016, 138, 8497–8504 CrossRef CAS PubMed.
- J. Hu, O. Adebali, S. Adar and A. Sancar, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6758–6763 CAS.
- B. A. Flusberg, D. R. Webster, J. H. Lee, K. J. Travers, E. C. Olivares, T. A. Clark, J. Korlach and S. W. Turner, Nat. Methods, 2010, 7, 461 CrossRef CAS PubMed.
- A. M. Fleming, Y. Ding and C. J. Burrows, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2604–2609 CrossRef CAS PubMed.
- G. P. Pfeifer, Int. J. Mol. Sci., 2018, 19, 1166 CrossRef PubMed.
- A. J. Likhachev, M. N. Ivanov, H. Bresil, G. Planche-Martel, R. Montesano and G. P. Margison, Cancer Res., 1983, 43, 829–833 CAS.
- S. Arimoto-Kobayashi, K. Kaji, G. M. A. Sweetman and H. Hayatsu, Carcinogenesis, 1997, 18, 2429–2433 CrossRef CAS.
- R. P. Jones, P. A. Sutton, J. P. Evans, R. Clifford, A. McAvoy, J. Lewis, A. Rousseau, R. Mountford, D. McWhirter and H. Z. Malik, Br. J. Cancer, 2017, 116, 923–929 CrossRef CAS PubMed.
- H. L. Gahlon, B. W. Schweizer and S. J. Sturla, J. Am. Chem. Soc., 2013, 135, 6384–6387 CrossRef CAS PubMed.
- H. L. Gahlon, M. L. Boby and S. J. Sturla, ACS Chem. Biol., 2014, 9, 2807–2814 CrossRef CAS PubMed.
- H. L. Gahlon and S. J. Sturla, Chem. – Eur. J., 2013, 19, 11062–11067 CrossRef CAS PubMed.
- L. A. Wyss, A. Nilforoushan, D. M. Williams, A. Marx and S. J. Sturla, Nucleic Acids Res., 2016, 44, 6564–6573 CrossRef PubMed.
- S. Obeid, A. Schnur, C. Gloeckner, N. Blatter, W. Welte, K. Diederichs and A. Marx, ChemBioChem, 2011, 12, 1574–1580 CrossRef CAS PubMed.
- C. Gloeckner, K. B. M. Sauter and A. Marx, Angew. Chem., Int. Ed., 2007, 46, 3115–3117 CrossRef CAS PubMed.
- K. Betz, A. Nilforoushan, L. A. Wyss, K. Diederichs, S. J. Sturla and A. Marx, Chem. Commun., 2017, 53, 12704–12707 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc00278b |
| This journal is © The Royal Society of Chemistry 2019 |