
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient visible light-driven water oxidation catalysed by an iron(IV) clathrochelate complex†
Sergii I.
Shylin
 *a,
Mariia V.
Pavliuk
a,
Luca
D’Amario
a,
Fikret
Mamedov
a,
Jacinto
Sá
a,
Gustav
Berggren
a and
Igor O.
Fritsky
*b
*a,
Mariia V.
Pavliuk
a,
Luca
D’Amario
a,
Fikret
Mamedov
a,
Jacinto
Sá
a,
Gustav
Berggren
a and
Igor O.
Fritsky
*b
aDepartment of Chemistry – Ångström Laboratory, Uppsala University, PO Box 523, 75120 Uppsala, Sweden. E-mail: sergii.shylin@kemi.uu.se
bDepartment of Chemistry, Taras Shevchenko National University of Kyiv, Volodymyrska 64, 01601 Kiev, Ukraine. E-mail: ifritsky@univ.kiev.ua
First published on 18th February 2019
Abstract
A water-stable FeIV clathrochelate complex catalyses fast and homogeneous photochemical oxidation of water to dioxygen with a turnover frequency of 2.27 s−1 and a maximum turnover number of 365. An FeV intermediate generated under catalytic conditions is trapped and characterised using EPR and Mössbauer spectroscopy.
Water oxidation powered by sunlight, preferably at ambient temperature and fairly neutral pH, is the main bottleneck for direct conversion of solar energy into molecular hydrogen as a solar fuel through photochemical water splitting.1 Ruthenium and iridium oxides are well-known heterogeneous water oxidation catalysts (WOCs).2 The most efficient homogeneous WOCs rely on complexes of the same noble metals.3 Due to the scarcity of Ru and Ir, there is considerable interest in the use of cheap base metal complexes for water oxidation. Extensive efforts have been devoted to developing homogeneous WOCs based on cobalt,4 manganese,5 copper,6 nickel,7 and iron.8 Among these metals, iron is arguably the most desirable candidate for catalyst development since it is the most abundant and least toxic.
Collins et al. were the first to demonstrate the potential of iron complexes as WOCs.9 They employed FeIII–TAML (TAML = tetraamido macrocyclic ligand) as a catalyst in chemical water oxidation to dioxygen by CeIV at pH 1 with a turnover number (TON) of 18 and turnover frequency (TOF) of 1.3 s−1. Later studies showed that iron complexes bearing polydentate N-donor ligands could catalyse oxidation of water with TONs over 100 in acidic media.10 However, the eventual goal is to develop efficient solar light-driven WOCs in order to circumvent the need for thermal and/or electrochemical oxidation. The photocatalytic water oxidation with [Ru(bpy)3]2+ as a photosensitiser and persulfate (S2O82−) as an electron acceptor is a common protocol to study the catalytic reactivity of WOCs.11 Following this approach, Chen et al. reported photochemical water oxidation using a number of simple FeII and FeIII complexes at pH 7–9.12 However, under oxidising conditions, Fe2O3 particles were produced and acted as the actual WOC. In an effort to prevent rapid ligand oxidation during photocatalysis, Panda et al. designed the biuret-modified FeIII–TAML, which showed improved stability over a wide pH range.13 The complex performed water oxidation with a maximum TON = 220 and TOF = 0.76 s−1 at pH 8.7. In contrast to precatalysts giving Fe2O3, the reversible formation of the FeV species as a possible intermediate during photocatalysis was reported. Moderate photocatalytic activity (maximum TON = 43.5; TOF = 0.6 s−1) at pH 8 has also been reported for molecular FeII pentapyridyl complexes in a study highlighting the importance of the sixth ligand on the stability of FeIV intermediates.14 In summary, several studies suggest the involvement of high-valent iron compounds (FeIV and FeV) as reactive transient species in the catalytic cycles of low-valent iron-based WOCs. Still, the catalytic activity of high-valent complexes represented mainly by oxo, nitrido and imido compounds is limited due to their low stability in aqueous solutions.15
We have recently reported a new class of high-valent iron complexes in which FeIV is stabilised by the hexahydrazide clathrochelate ligand.16 The complexes are spontaneously assembled in aqueous media from Fe salts, oxalodihydrazide and formaldehyde in the presence of atmospheric oxygen. These cage complexes are exceptionally stable in water under ambient conditions, thus suggesting a potential as WOCs. In this communication, we report the photochemical water oxidation catalysed by Na2[FeIV(L-6H)]·2H2O (1), where L is the ligand constructed by linking three oxalodihydrazide residues with six methylene bridges. Mössbauer, EPR spectroscopy and DLS experiments show that the molecular catalyst remains intact during water oxidation and no low-valent iron species (e.g., oxides) are formed. We also report the photochemical generation of an FeV (S = 1/2) complex, and this proposed oxidised intermediate is characterised using ESI-MS, EPR, UV-Vis, and Mössbauer spectroscopy.
Complex 1 was prepared according to the previously described procedure16 with minor modifications (ESI†). Its hyperfine parameters (isomer shift δ = 0.118(2) mm s−1 and quadrupole splitting |ΔEQ| = 2.493(3) mm s−1) derived from the Mössbauer spectrum (Fig. S1, ESI†) corroborate the intermediate spin state (S = 1) of tetravalent iron,15a–c whereas the presence of other iron species in the sample is ruled out. The cyclic voltammograms (CV) of 1 carried out in borate buffer at pH 8.0 (under similar conditions to those used for water oxidation studies) display a quasi-reversible feature at E1/2 = +0.92 V versus a normal hydrogen electrode (NHE) corresponding to the FeV/FeIV couple (Fig. S2, ESI†). This is followed by an oxidative process starting at +1.15 V versus NHE that we assign to water oxidation (Fig. 1a). For the anodic peak, the current Ip is linearly dependent on v1/2 (where v is the scan rate in mV s−1) (Fig. 1b) which indicates a diffusion process in solution. The onset of the catalytic wave appears at an overpotential of 0.39 V, which is relatively mild compared to other iron-based catalysts.13,14,17 The stability of 1 under electrocatalytic conditions was verified by UV-Vis spectroscopy on samples collected after 2 h of sustained electrolysis (Fig. S3, ESI†). Importantly, catalytic water oxidation in the presence of 1 is thermodynamically accessible by [Ru(bpy)3]3+ (E1/2 = +1.26 V versus NHE) that can be generated from the light-excited state of [Ru(bpy)3]2+ by sacrificial oxidants.
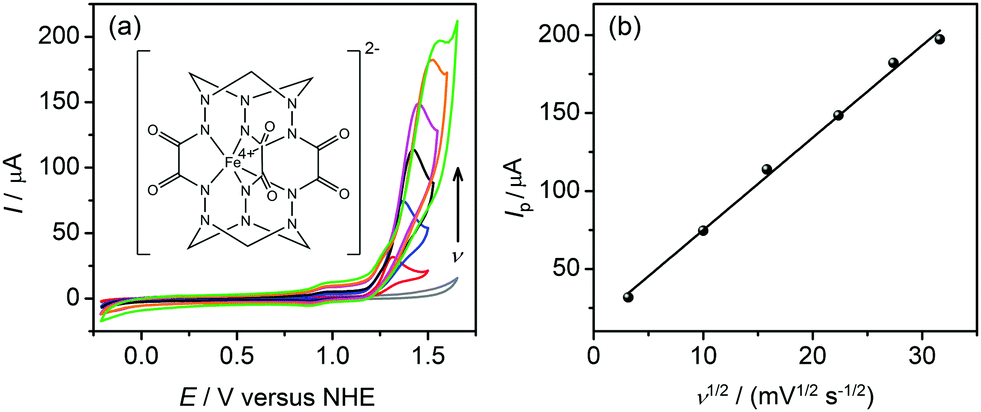 | ||
| Fig. 1 (a) Cyclic voltammograms of 1 (0.25 mM) at pH 8.0 recorded at scan rates v from 10 to 1000 mV s−1, and background (grey line). The molecular structure of 1 is inserted. (b) The peak current Ip as a function of v1/2 for catalytic water oxidation. | ||
The activity of 1 in photochemical water oxidation was evaluated using the molecular photosensitiser [Ru(bpy)3](ClO4)2 and an excess amount of electron acceptor Na2S2O8 in a buffered aqueous solution. At a concentration of 1 = 0.5 μM, oxygen evolution was observed with a TON = 365 using a standard Clark electrode (Fig. 2a). Subsequent kinetic experiments revealed that the initial rates of oxygen evolution had a linear dependence on the catalyst concentration (Fig. 2b and Fig. S4, ESI†), indicating a pseudo-first order reaction. From this, a TOF = 2.27(6) s−1 can be derived that reflects considerably higher reaction rates with respect to other iron-based molecular WOCs.13,14 The pH of the reaction mixture was found to have only a minor effect on oxygen evolution in the range 7.0–9.2, although 1 showed the highest performance at pH 8.0 (Fig. S5, ESI†). Chemical water oxidation using [Ru(bpy)3]3+ was carried out with catalyst 1 to confirm that O2 is evolved from water rather than the sacrificial electron acceptor. Also, under these conditions, efficient H2O oxidation was observed with a TOF of 3.3(1) s−1 (Fig. S6, ESI†). Moreover and in contrast to the photochemical assay, when O2 was detected after a 2–3 s lag phase, which was needed to build up [Ru(bpy)3]3+, catalysis started immediately if 1 was mixed with chemically prepared [Ru(bpy)3]3+.
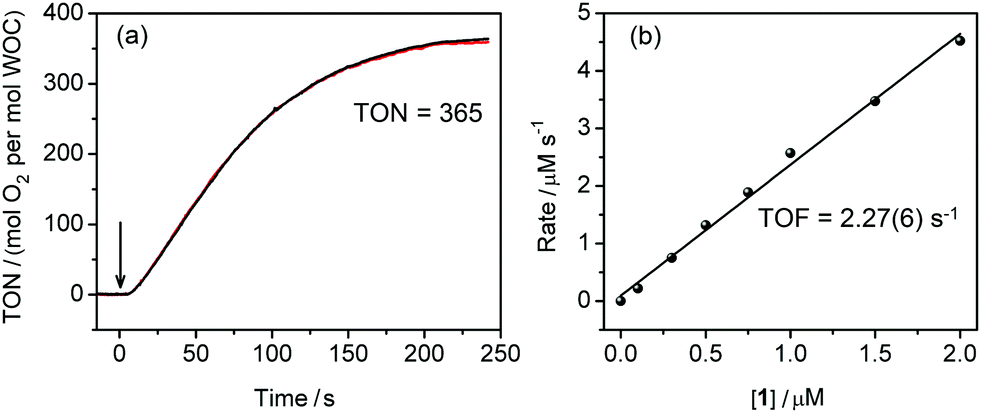 | ||
| Fig. 2 (a) Plot of O2 evolution for the photocatalytic system consisting of 1 (0.5 μM), [Ru(bpy)3](ClO4)2 (0.2 mM), and Na2S2O8 (2 mM) at pH 8.0 reproduced in two experiments (black and red curves). The arrow indicates start of irradiation with LEDs (λ = 450 nm). (b) Initial rate of O2 formation versus concentration of 1. | ||
To elucidate the role of 1 in water oxidation, a series of UV-Vis spectroscopy experiments was performed. Irradiation of the mixture of [Ru(bpy)3]2+ and S2O82− at 450 nm resulted in bleaching of the 452 nm [Ru(bpy)3]2+ band and appearance of the broad [Ru(bpy)3]3+ band at 670 nm (Fig. S7, ESI†). When 1 was added, no accumulation of photogenerated RuIII could be observed in the steady-state spectra suggesting its rapid quenching by the WOC. To estimate the rate of this process, time-resolved transient absorption (TA) measurements were performed. The microsecond TA spectra of the catalytic system selectively excited by a 460 nm nanosecond pulse showed a characteristic [Ru(bpy)3]2+ bleach at ∼452 nm (Fig. 3a), whose intensity decreased with time. The trace of the optical density (OD) at 420 nm showed a mono-exponential decay of the bleach with a lifetime τ = 15(1) μs (Fig. 3b, red trace). In contrast, the trace of the OD for the solution without 1 demonstrated no reduction of [Ru(bpy)3]3+ (Fig. 3b, blue trace) pointing to the hole-scavenging activity of the WOC.
 | ||
| Fig. 3 (a) Transient absorption spectra showing photoinduced bleaching of the [Ru(bpy)3]2+ band in the presence of Na2S2O8 and 1. (b) Kinetic traces at 420 nm for the solutions containing [Ru(bpy)3]2+ and Na2S2O8 in the absence (blue) and presence (red, fit shown in black) of 1 showing quenching of [Ru(bpy)3]3+ by 1. | ||
When the hole is injected from [Ru(bpy)3]3+ to 1, oxidation of the complex molecule and appearance of a new species, which is characterised by a broad absorption at around 835 nm in the UV-Vis spectrum, can be observed (Fig. 4a). Based on EPR and Mössbauer spectroscopy studies at low temperatures (see below) and ESI-MS experiments (Fig. S8, ESI†), we attribute this species to the FeV intermediate with composition [FeV(L-6H)]−. However, it is unstable under ambient conditions and rapidly reduced at room temperature (Fig. 4b). This leads to complete regeneration of FeIV and in 20 minutes the spectrum of the reaction mixture is virtually identical to that before irradiation. The FeV intermediate can be generated repeatedly by irradiation at 450 nm that underscores the stability of 1. The FeV species is also obtained by direct oxidation of 1 using freshly prepared [Ru(bpy)3]3+ (Fig. S9, ESI†). The subsequent regeneration of FeIV is accompanied by slow oxygen evolution (Fig. S10, ESI†). Still, this process is arguably too slow to be the main contributing process in fast catalysis. In agreement with this, our CV data suggest that oxidation beyond the FeV state is needed to generate a significant catalytic current.
 | ||
| Fig. 4 (a) The UV-Vis spectra of a solution containing 1, [Ru(bpy)3](ClO4)2, and Na2S2O8 at pH 8.0 before illumination (black), directly after illumination at 450 nm (red), and in 20 minutes after illumination (blue). Evolution of the spectra is shown in grey. (b) Decay of the absorption at 830 nm from the photogenerated FeV species. | ||
EPR spectroscopy was used to characterise possible intermediates involved in photochemical water oxidation. Complex 1 as-prepared was found to be EPR-silent down to 5 K, characteristic of FeIV with S = 1. In order to probe the system under photocatalytic conditions, 1 was added to a buffered aqueous solution containing [Ru(bpy)3]2+ (S = 0) and S2O82−. The mixture was then illuminated at 450 nm during 20 s followed by immediate freezing. This allowed us to trap and observe a newly generated species characterised by a relatively broad EPR signal with g ≈ 2.0 (Fig. 5a, red).18 It most likely originates from the intermediate detected in the electronic spectra and can be assigned to low-spin (S = 1/2) FeV, based on signal shape and its power saturation behaviour (Fig. S11, ESI†). As [Ru(bpy)3]3+ (S = 1/2) can be easily identified by a characteristic pattern (Fig. 5a, blue), its presence in the photochemical reaction mixture is ruled out. Our EPR findings are complemented by Mössbauer spectroscopy of the frozen solution. The Mössbauer spectrum recorded at 80 K can be fitted with two symmetrical doublets with relative intensities of 72(3)% (doublet I) and 28(3)% (doublet II) (Fig. 5b). The hyperfine parameters of doublet I (δ = 0.12(2) mm s−1 and |ΔEQ| = 2.42(4) mm s−1) match those for pure 1 in the solid state. Doublet II has significantly lower δ = −0.03(2) mm s−1 indicating metal-centred oxidation resulting in the formation of FeV species.15b,19 The higher |ΔEQ| = 3.32(5) mm s−1 for doublet II originates from the appearance of a negative valence contribution to the electric field gradient due to withdrawing one electron from the dx2−y2/dxy level. Hence, Mössbauer spectroscopy unambiguously shows the presence of the FeV intermediate with the electronic configuration (dz2)2(dx2−y2/dxy)1 in the reaction mixture.
 | ||
| Fig. 5 (a) The EPR spectra of the frozen solution (5 K) containing 1, [Ru(bpy)3](ClO4)2, and Na2S2O8 before (black) and after illumination at 450 nm (red). The spectrum of photochemically generated RuIII is given for comparison (blue). (b) The 57Fe Mössbauer spectrum (80 K) showing oxidation of FeIV to FeV under photocatalytic conditions. | ||
As certain iron-based complexes are often claimed as precatalysts, which give catalytically active hematite nanoparticles,11a,12 we performed DLS studies to probe the molecular integrity of 1. Upon irradiation of the catalytic system at pH 8.0, no evidence for particle formation was found. In contrast, when 1 is replaced by Fe(ClO4)3 in our assay mixture, particles are formed immediately (Fig. S12, ESI†). Our spectroscopy studies also corroborate the stability of 1. First, the UV-Vis spectra show that FeIV can be oxidised to FeV and then quantitatively reduced back to FeIV with no signs of decomposition. Second, the fast quenching of [Ru(bpy)3]3+ speaks in favour of the molecular nature of the hole acceptor. Third, as a local sensitive probe, Mössbauer spectroscopy does not show appearance of any low-valent iron species in the reaction mixture. Intermediates beyond FeV appear to be too active and have so far eluded detection in the steady-state spectra. As the Fe ion in 1 is encapsulated in the robust clathrochelate ligand, formation of a typical oxoiron active species would require a seven-coordinated Fe intermediate.20 However, we cannot exclude the involvement of the ligand in the O–O bond formation, as has been suggested for molecular Ru-based WOCs.21 A combined experimental and computational study of the mechanism of (photo-) chemical water oxidation by the clathrochelate complex 1 is currently on-going.
This study introduces the high-valent iron complex 1 as an efficient WOC exhibiting >300 turnovers in combination with a relatively low water oxidation overpotential. To the best of our knowledge, it is the highest TON observed for mononuclear iron complexes in photochemical water oxidation. The synthetic versatility of clathrochelate complexes provides one avenue to fine tune their redox and catalytic properties.
This work was supported by the Swedish Institute grant awarded to SIS and Horizon 2020 MSCA-RISE-2017 grant No 778245.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For selected reviews on artificial photosynthesis, see:
(a) J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455 CrossRef CAS PubMed
; (b) N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed
-
(a) C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed
-
(a) L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418 CrossRef CAS PubMed
-
(a) T. Nakazono, A. R. Parent and K. Sakai, Chem. Commun., 2013, 49, 6325 RSC
-
(a) M. M. Najafpour, G. Renger, M. Hołyńska, A. N. Moghaddam, E.-M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J.-R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886 CrossRef CAS PubMed
-
(a) S. J. Koepke, K. M. Light, P. E. VanNatta, K. M. Wiley and M. T. Kieber-Emmons, J. Am. Chem. Soc., 2017, 139, 8586 CrossRef CAS PubMed
-
(a) J.-W. Wang, W.-L. Liu, D.-C. Zhong and T.-B. Lu, Coord. Chem. Rev., 2019, 378, 237 CrossRef CAS
-
(a) M. Okamura, M. Kondo, R. Kuga, Y. Kurashige, T. Yanai, S. Hayami, V. K. K. Praneeth, M. Yoshida, K. Yoneda, S. Kawata and S. Masaoka, Nature, 2016, 530, 465 CrossRef CAS PubMed
- W. C. Ellis, N. D. McDaniel, S. Bernhard and T. J. Collins, J. Am. Chem. Soc., 2010, 132, 10990 CrossRef CAS PubMed
-
(a) Z. Codolà, I. Garcia-Bosch, F. Acuña-Parés, I. Prat, J. M. Luis, M. Costas and J. Lloret-Fillol, Chem. – Eur. J., 2013, 19, 8042 CrossRef PubMed
-
(a) S. Fukuzumi, J. Jung, Y. Yamada, T. Kojima and W. Nam, Chem. – Asian J., 2016, 11, 1138 CrossRef CAS PubMed
- G. Chen, L. Chen, S.-M. Ng, W.-L. Man and T.-C. Lau, Angew. Chem., Int. Ed., 2013, 52, 1789 CrossRef CAS PubMed
- C. Panda, J. Debgupta, D. Díaz Díaz, K. K. Singh, S. Sen Gupta and B. B. Dhar, J. Am. Chem. Soc., 2014, 136, 12273 CrossRef CAS PubMed
- B. Das, A. Orthaber, S. Ott and A. Thapper, ChemSusChem, 2016, 9, 1178 CrossRef CAS PubMed
- For selected examples of high-valent iron complexes, see:
(a) J. England, J. O. Bigelow, K. M. Van Heuvelen, E. R. Farquhar, M. Martinho, K. K. Meier, J. R. Frisch, E. Münck and L. Que Jr., Chem. Sci., 2014, 5, 1204 RSC
- S. Tomyn, S. I. Shylin, D. Bykov, V. Ksenofontov, E. Gumienna-Kontecka, V. Bon and I. O. Fritsky, Nat. Commun., 2017, 8, 14099 CrossRef CAS PubMed
-
(a) G. Panchbhai, W. M. Singh, B. Das, R. T. Jane and A. Thapper, Eur. J. Inorg. Chem., 2016, 3262 CrossRef CAS
-
(a) F. Tiago de Oliveira, A. Chanda, D. Banerjee, X. Shan, S. Mondal, L. Que Jr, E. L. Bominaar, E. Münck and T. J. Collins, Science, 2007, 315, 835 CrossRef CAS PubMed
- A. Chanda, D.-L. Popescu, F. Tiago de Oliveira, E. L. Bominaar, A. D. Ryabov, E. Münck and T. J. Collins, J. Inorg. Biochem., 2006, 100, 606 CrossRef CAS PubMed
- Q. Daniel, P. Huang, T. Fan, Y. Wang, L. Duan, L. Wang, F. Li, Z. Rinkevicius, F. Mamedov, M. S. G. Ahlquist, S. Styring and L. Sun, Coord. Chem. Rev., 2017, 346, 206 CrossRef CAS
- J. K. Hurst, J. L. Cape, A. E. Clark, S. Das and S. Qin, Inorg. Chem., 2008, 47, 1753 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplementary figures. See DOI: 10.1039/c9cc00229d |
| This journal is © The Royal Society of Chemistry 2019 |