
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A rhodium-catalysed Sonogashira-type coupling exploiting C–S functionalisation: orthogonality with palladium-catalysed variants†
Milan
Arambasic
,
Manjeet K.
Majhail
,
Robert N.
Straker
 ,
James D.
Neuhaus
and
Michael C.
Willis
*
,
James D.
Neuhaus
and
Michael C.
Willis
*
Department of Chemistry, University of Oxford, Chemistry Research Laboratory, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: michael.willis@chem.ox.ac.uk
First published on 14th February 2019
Abstract
This report concerns the development of an efficient Sonogashira-type coupling of arylmethylsulfides and terminal alkynes to generate aryl alkyne motifs. Orthogonal reactivity between traditional Pd catalysts, and the Rh catalysts employed, results in the ability to selectively activate either the C–S bond or C–X bond through catalyst choice. The Rh–bisphosphine catalyst has further been shown to be able to effect a hydroacylation-Sonogashira tandem sequence, and in combination with further onward reactions has been used in the synthesis of heterocycles and polycyclic systems.
Since its discovery in 1975 by the laboratories of Cassar, Heck and Sonogashira,1 the coupling of aryl halides with terminal alkynes has represented one of the most popular methods of assembling aryl alkynes.2 The prototypical “Sonogashira coupling” revolves around the co-operative action of a Pd-catalyst with a Cu-co-catalyst, used to activate the terminal alkyne. Synthetically straightforward, the reaction has been the subject of countless studies looking to expand the scope and efficiency of the process. Included amongst these developments are studies aimed at developing Cu-free processes (in order to reduce Cu-catalyzed Glaser-type alkyne homo-dimerization, and to reduce the environmental impact),3 transition metal-free processes,4 and expanding the range of, and consequently selectivity between, leaving groups.2 Whilst simple aryl/alkenyl iodides, bromides, chlorides and triflates have provided a plethora of examples under a number of reaction conditions, selectivity for reaction with one halide/pseudohalide over another has not proven straightforward. Use of a Ni-catalyst by Hu and co-workers in 2009 has shown promising levels of selectivity by exploiting the innate reactivity of alkyl chlorides, bromides and iodides; however, this work did not deal with aryl substitution, and other than by redesigning the substrate, there is no way to reverse the selectivity.5 A number of laboratories have looked at using C–H functionalisation chemistry to overcome this problem with some success, however, the use of simple terminal alkynes as the coupling partner remains rare,6 with haloalkynes,7 or hypervalent iodine reagents8 being the reagents of choice in “inverse Sonogashira coupling reactions”.
The activation of carbon–sulfur bonds, an area of interest in the Willis laboratory for a number of years, has seen significant expansion beyond the simple Kumada coupling in recent decades.9 Reactions such as the Liebeskind–Srogl coupling are now regularly employed in both academic and industrial laboratories.10 Many of these processes employ catalysts not known for their reactivity with aryl halides, and as a result a number of orthogonal processes have been developed, almost exclusively on highly activated heteroaromatic scaffolds.11
Reports of desulfitative alkynylation are rare, and involve activation of the sulfide component,12 metallated alkynes,13 or are the result of the coupling of alkynyl thioethers with organometallic reagents.14 Our laboratory has recently demonstrated that, in combination with a suitable directing group, it is possible to use Rh–bisphosphine complexes to selectively activate the C–S bond of arylmethylsulfides, to effect carbothiolation reactions,15 reductive desulfurisations,16 or Suzuki-type cross coupling reactions (Scheme 1).17 As full selectivity for C–S functionalisation over C–X functionalisation was in each case observed, we envisaged that similar methodology would enable us to develop an orthogonal Sonogashira-type process simply by switching the boronic acid coupling partner with a terminal alkyne.
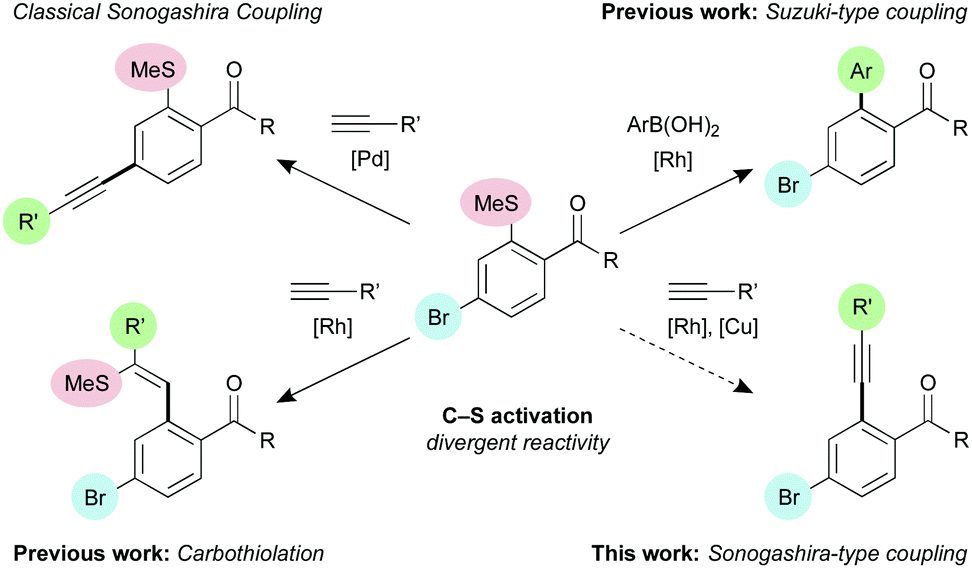 | ||
| Scheme 1 Existing C–S activation methodologies and proposed chemoselective Sonogashira-type coupling. | ||
Whilst terminal alkynes had previously been used as the coupling partner in the carbothiolation procedure, we anticipated that the use of a thiophilic Cu-co-catalyst would favor the alkynylation over carbothiolation, thus providing an example of catalyst-controlled divergence of reactivity. Initial investigations involved the combination of 2-(methylthio)acetophenone (1a) and phenylacetylene (2a). By using the hemilabile bisphosphine ligand DPEphos in combination with [Rh(nbd)2]BF4, copper iodide and potassium carbonate (Table 1, entry 1), a conversion of 25% was recorded to product 3a. By switching to the more rigid XantPhos ligand, the conversion dropped to 13% and the product of carbothiolation (4) was also observed (entry 2). Moving towards small bite-angle PCP-bisphosphines, such as dppm and dcpm (entries 3 and 4), did not show much improvement, however, with the PCCP ligands dppe and dcpe (entries 5 and 6, respectively), considerably higher conversions were achieved. Whilst the conversion was lower with dcpe than with dppe, the improved selectivity made it an attractive choice for further optimization. After evaluating a number of bases, it was found that silver carbonate could improve the conversion to 61% (entry 7). Experimenting with various copper sources (entries 8–11) resulted in conversions of up to 93%; however, the breakthrough came with the use of copper bromide (entry 12). Not only was full consumption of the starting material now observed, but only product 3a was observed in the 1H-NMR spectrum of the crude reaction mixture. This high conversion and selectivity was reflected in the excellent isolated yield of 95%. Aware that the use of stoichiometric silver carbonate could be costly on a larger scale, alternative conditions employing potassium carbonate were also developed (entry 13).
With efficient conditions for the formation of Sonogashira-product 3a in hand, we moved on to investigate the range of terminal alkynes compatible with this chemistry (Scheme 2). Aromatic alkynes proved excellent substrates, with high yields observed for both electron-rich (3b) and electron-poor (3c and d) aryl groups, as well as heteroaromatic alkynes (3e). As anticipated, the Rh-complex exhibited high selectivity for reaction with the carbon–sulfur bond over that of the carbon–halide bond present in 3c. Alkyl alkynes performed more poorly with the standard substrate (3f–i), however we were pleased to observe that with alternative directing groups, excellent yields were still possible (3g). Sterically bulky groups such as tert-butyl (3h) or the removable TIPS group (3j) were well tolerated. By using conditions A, it was possible to perform the reaction on a 30.0 mmol scale, using only 2.5 mol% of the active rhodium catalyst, delivering diaryl alkyne 3a in a 96% yield.
|
|
||||
|---|---|---|---|---|
| Entry | Ligand | Cu source | Additive | Conversiona |
| a Conversions determined by 1H NMR spectroscopic analysis of the crude reaction mixture, value in parentheses indicates conversion to carbothiolation product 4. b Isolated yield of 3a. | ||||
| 1 | DPEphos | CuI | K2CO3 | 25% |
| 2 | Xantphos | CuI | K2CO3 | 13% (5%) |
| 3 | dppm | CuI | K2CO3 | 19% (5%) |
| 4 | dcpm | CuI | K2CO3 | 30% |
| 5 | dppe | CuI | K2CO3 | 48% (7%) |
| 6 | dcpe | CuI | K2CO3 | 40% |
| 7 | dcpe | CuI | Ag2CO3 | 61% (12%) |
| 8 | dcpe | CuI (2 eq.) | Ag2O | 84% (8%) |
| 9 | dcpe | CuI (2 eq.) | Ag2CO3 | 93% (5%) |
| 10 | dcpe | CuTc | Ag2CO3 | — |
| 11 | dcpe | Cu(OAc) | Ag2CO3 | — |
| 12 | dcpe | CuBr (2 eq.) | Ag2CO3 | 100%, 95%b |
| 13 | dcpe | CuBr (4 eq.) | K2CO3 (2 eq.) | 100% |

 | ||
| Scheme 2 Scope of alkyne component in Sonogashira-type coupling reactions. | ||
As shown in Scheme 3, exploration of the scope of the arylmethylsulfide component was similarly successful, with electron-rich (3k) and electron-poor (3l–o) examples each reacting in good to excellent yields. The generation of halide containing products allows for further functionalisation. It was additionally demonstrated that more complex functionality could be tolerated elsewhere on the scaffold (3p).
 | ||
| Scheme 3 Scope of arylmethylsulfide component. | ||
As previously mentioned, a major strength of rhodium(I) catalysis lies in the extremely high selectivity for the activation of one bond, in this case the C–S bond, in the presence of other easily activated bonds, which could not be carried through a reaction sequence if using palladium catalysis, for example. In order to demonstrate the orthogonality of these processes, we first exposed compound 5, featuring both aryl bromide and methylsulfide functional groups, to our Rh(I)-catalyzed C–S Sonogashira conditions, followed by traditional Pd(0)-catalyzed Sonogashira conditions for the generation of dialkyne 7 (Scheme 4). Each reaction proceeded in a very good yield, and the same product (7), was obtained by switching the order of the two transition metal-catalyzed processes.
Our laboratory has previously demonstrated that in situ generated Rh–dcpe complexes are effective catalysts for alkene and alkyne hydroacylation reactions,18 and we were therefore intrigued by the possibility of conducting a tandem hydroacylation-Sonogashira sequence, resulting in the three-component coupling of an aldehyde with two distinct alkynes, forming two new C–C bonds, using a single rhodium catalyst (Scheme 5). Such a process would be a powerful method to rapidly build up molecular complexity from simple, commercially available building blocks. We began by reducing potential complications by using the same alkyne for both steps of the reaction, with fluoro-substituted phenylacetylene affording the product (9a) in good yield over two steps. We were pleased to discover that the use of differing alkynes for each step was also easily achieved. A range of alkynes was employed, generating hydroacylation-Sonogashira products 9b–g. Compound 9g, in particular, contains synthetic handles for further functionalisation on each of the constituent components, which would be extremely difficult to prepare using Pd- or Ni-catalyzed methods.
 | ||
| Scheme 4 Reversible selectivity between Rh- and Pd-catalyzed Sonogashira reactions. | ||
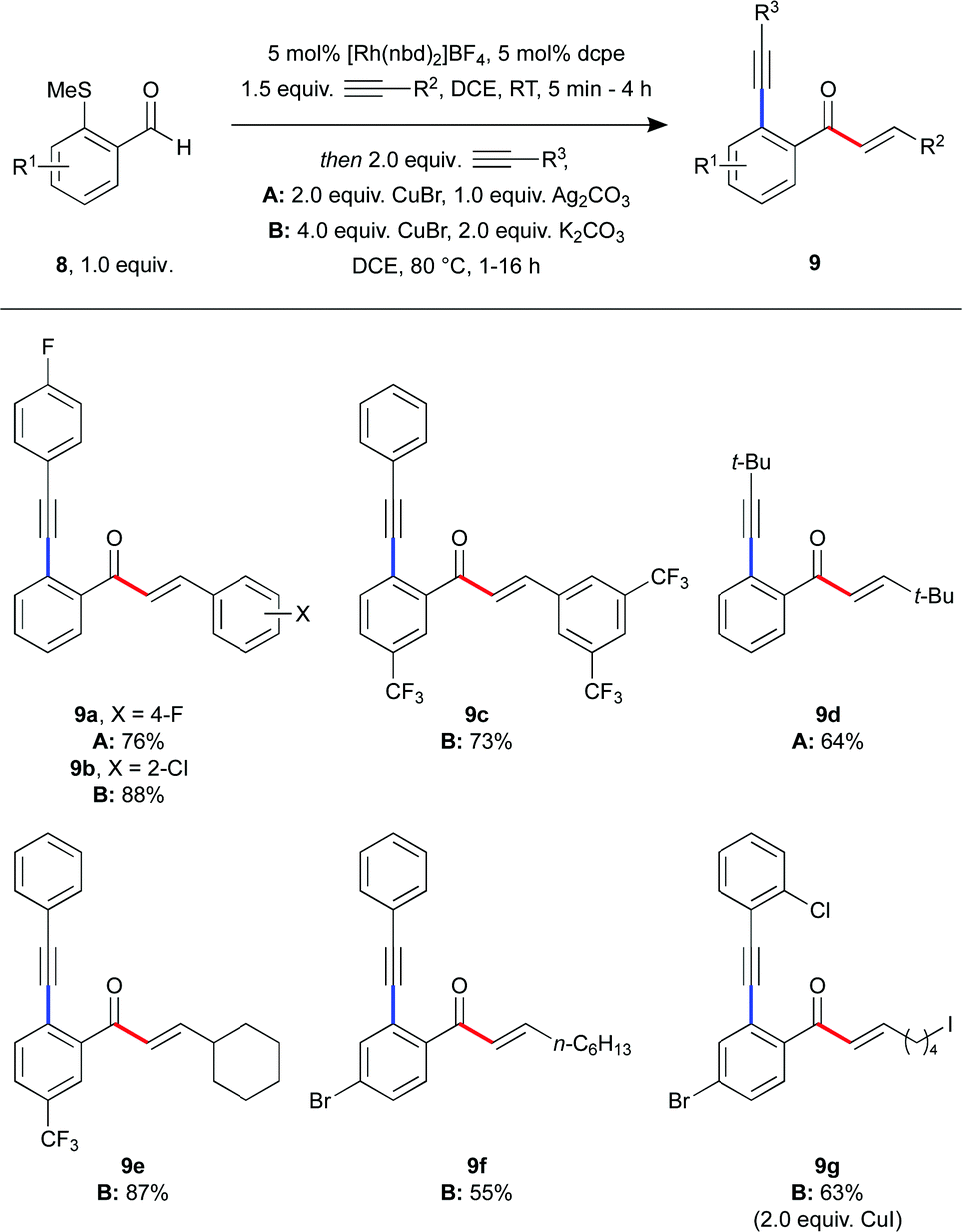 | ||
| Scheme 5 Tandem hydroacylation-Sonogashira sequence. | ||
Further reaction sequences, generating increasingly complex products were subsequently attempted, as depicted in Scheme 6. By employing suitably protected propargyl amines as substrates for the first step in the hydroacylation-Sonogashira sequence,19 a three-step, two-pot synthesis of highly functionalised pyrrole 11 was achieved in a good yield, requiring only a single chromatographic purification to obtain the pure heterocycle. An alternative sequence resulted in the combination of the hydroacylation-Sonagashira steps with a π-acid-catalysed [4+2]-cycloaddition reaction between an isochromenylium ion and a styrene derivate, ultimately yielding 1,2-dihydronaphthylene 13 after subsequent ring opening of the bicyclic intermediate. Such processes have been effected by a number of π-acids, and the conditions used herein were first reported by Yamamoto et al. in 2003.20
 | ||
| Scheme 6 Tandem reactions for the synthesis of heterocycles and extended ring systems. | ||
In conclusion, we have developed a novel Sonogashira-type coupling between arylmethylsulfides and terminal alkynes. Utilizing an electrophilic Rh–bisphosphine catalyst system, the reaction has been shown to exhibit tolerance to a broad range of functional groups, many of which would not be tolerated under alternative TM-catalysis. This selectivity has been demonstrated in the orthogonal activation of either a C–S bond or C–X bond by either Rh or Pd respectively. In addition, the reactions reported here show how divergent reactivity for the same reaction components can be achieved by catalyst and reagent selection, with Sonogashira and not carbothiolation products being obtained.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) K. Sonogashira, Y. Tohda and N. Hagihara, Tetrahedron Lett., 1975, 16, 4467 CrossRef; (b) L. Cassar, J. Organomet. Chem., 1975, 93, 253 CrossRef CAS; (c) H. A. Dieck and F. R. Heck, J. Organomet. Chem., 1975, 93, 259 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed.
- (a) V. P. W. Böhm and W. A. Hermmann, Eur. J. Org. Chem., 2000, 3679 CrossRef; (b) B. H. Lipshutz, D. W. Chung and B. Rich, Org. Lett., 2008, 10, 3793 CrossRef CAS PubMed; (c) L. Dong-Hwan, H. Qiu, M.-h. Cho, I.-M. Lee and M.-J. Jin, Synlett, 2008, 1657 Search PubMed; (d) Y. Liang, Y.-X. Xie and J.-H. Li, J. Org. Chem., 2006, 71, 379 CrossRef CAS PubMed; (e) V. K. Kanuru, S. M. Humphrey, J. M. W. Kyffin, D. A. Jefferson, J. W. Burton, M. Armbrüster and R. M. Lambert, Dalton Trans., 2009, 7602 RSC.
- (a) P. Appukkuttan, W. Dehaen and E. Van der Eycken, Eur. J. Org. Chem., 2003, 4713 CrossRef CAS; (b) N. E. Leadbeater, M. Marco and B. J. Tominack, Org. Lett., 2003, 5, 3919 CrossRef CAS PubMed; (c) M. S. Maji, S. Murarka and A. Studer, Org. Lett., 2010, 12, 3878 CrossRef CAS PubMed.
- O. Vechorkin, D. Barmaz, V. Proust and X. Hu, J. Am. Chem. Soc., 2009, 131, 12078 CrossRef CAS PubMed.
- M. Shang, H.-L. Wang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 11590 CrossRef CAS PubMed.
- (a) I. V. Seregin, V. Ryabova and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 7742 CrossRef CAS PubMed; (b) A. S. Dudnik and V. Gevorgyan, Angew. Chem., Int. Ed., 2010, 49, 2096 CrossRef CAS PubMed; (c) M. Tobisu, Y. Ano and N. Chatani, Org. Lett., 2009, 11, 3250 CrossRef CAS PubMed.
- (a) C. Feng and T.-P. Loh, Angew. Chem., Int. Ed., 2014, 53, 2722 CrossRef CAS PubMed; (b) F. Xie, Z. Qi, S. Yu and X. Li, J. Am. Chem. Soc., 2014, 136, 4780 CrossRef CAS PubMed.
- (a) L. Wang, W. He and Z. Yu, Chem. Soc. Rev., 2013, 42, 599 RSC; (b) S. Otsuka, K. Nogi and H. Yorimitsu, Top. Curr. Chem., 2018, 376 Search PubMed.
- (a) L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260 CrossRef CAS; (b) H. Prokopcová and C. O. Kappe, Angew. Chem., Int. Ed., 2009, 48, 2276 CrossRef PubMed.
- (a) A. Aguilar-Aguilar and E. Peña-Cabrera, Org. Lett., 2007, 9, 4163 CrossRef CAS PubMed; (b) C. Kusturin, L. S. Liebeskind, H. Rahman, K. Sample, B. Schweitzer, J. Srogl and W. L. Neumann, Org. Lett., 2003, 5, 4349 CrossRef CAS PubMed; (c) A. Tikad, S. Routier, M. Akssira, J.-M. Leger, C. Jarry and G. Guillaumet, Org. Lett., 2007, 9, 4673 CrossRef CAS PubMed; (d) V. P. Mehta, A. Sharma, K. Van Hecke, L. Van Meervelt and E. Van der Eycken, J. Org. Chem., 2008, 73, 2382 CrossRef CAS PubMed; (e) N. Kaval, B. K. Singh, D. S. Ermolat'ev, S. Claerhout, V. S. Parmar, J. Van der Eycken and E. Van der Eycken, J. Comb. Chem., 2007, 9, 446 CrossRef CAS PubMed.
- (a) V. P. Mehta, A. Sharma and E. Van der Eycken, Org. Lett., 2008, 10, 1147 CrossRef CAS PubMed; (b) Z.-Y. Tian, S.-M. Wang, S.-J. Jia, H.-X. Song and C.-P. Zhang, Org. Lett., 2017, 19, 5454 CrossRef CAS PubMed.
- A. Baralle, H. Yorimitsu and A. Osuka, Chem. – Eur. J., 2016, 22, 10768 CrossRef CAS PubMed.
- (a) L. Melzig, J. Stemper and P. Knochel, Synthesis, 2010, 2085 CAS; (b) C. Savarin, J. Srogl and L. S. Liebeskind, Org. Lett., 2001, 3, 91 CrossRef CAS PubMed.
- (a) J. F. Hooper, A. B. Chaplin, C. González-Rodríguez, A. L. Thompson, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 2906 CrossRef CAS PubMed; (b) M. Arambasic, J. F. Hooper and M. C. Willis, Org. Lett., 2013, 15, 5162 CrossRef CAS PubMed.
- J. F. Hooper, R. D. Young, A. S. Weller and M. C. Willis, Chem. – Eur. J., 2013, 19, 3125 CrossRef CAS PubMed.
- J. F. Hooper, R. D. Young, I. Pernik, A. S. Weller and M. C. Willis, Chem. Sci., 2013, 4, 1568 RSC.
- (a) A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885 CrossRef CAS PubMed; (b) T. J. Coxon, M. Fernández, J. Barwick-Silk, A. I. McKay, L. E. Britton, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2017, 139, 10142 CrossRef CAS PubMed; (c) R. N. Straker, M. K. Majhail and M. C. Willis, Chem. Sci., 2017, 8, 7963 RSC; (d) R. N. Straker, M. Formica, J. D. Lupton, J. Niu and M. C. Willis, Tetrahedron, 2018, 74, 5408 CrossRef CAS; (e) M. Castaing, S. L. Wason, B. Estepa, J. F. Hooper and M. C. Willis, Angew. Chem., Int. Ed., 2013, 52, 13280 CrossRef CAS PubMed.
- M. K. Majhail, P. M. Ylioja and M. C. Willis, Chem. – Eur. J., 2016, 22, 7879 CrossRef CAS PubMed.
- N. Asao, T. Kasahara and Y. Yamamoto, Angew. Chem., Int. Ed., 2003, 42, 3504 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc00092e |
| This journal is © The Royal Society of Chemistry 2019 |