
Mineral surfaces select for longer RNA molecules†
Ryo
Mizuuchi
 *a,
Alex
Blokhuis
bc,
Lena
Vincent
d,
Philippe
Nghe
b,
Niles
Lehman
a and
David
Baum
d
*a,
Alex
Blokhuis
bc,
Lena
Vincent
d,
Philippe
Nghe
b,
Niles
Lehman
a and
David
Baum
d
aDepartment of Chemistry, Portland State University, Portland, OR 97201, USA. E-mail: mizuuchi@pdx.edu
bLaboratory of Biochemistry, PSL Research University, ESPCI, 10 rue Vauquelin, F-75231 Paris Cedex 05, France
cGulliver Laboratory, UMR CNRS 7083, PSL Research University, ESPCI, 10 rue Vauquelin, F-75231 Paris Cedex 05, France
dDepartment of Botany, Wisconsin Institute for Discovery, University of Wisconsin-Madison, Madison, WI-53715, USA
First published on 17th January 2019
Abstract
We report empirically and theoretically that multiple prebiotic minerals can selectively accumulate longer RNAs, with selectivity enhanced at higher temperatures. We further demonstrate that surfaces can be combined with a catalytic RNA to form longer RNA polymers, supporting the potential of minerals to develop genetic information on the early Earth.
Mineral surfaces, ubiquitous on the Earth, likely played diverse roles in the origins of life,1 including in the proposed RNA world, when RNA molecules acted as both informational polymers and catalysts.2,3 It has been shown that minerals catalyze the synthesis of RNA components4,5 and RNA polymers,6,7 and can protect RNA from degradation.8,9 In 1980, Orgel noticed another potential of mineral surfaces: the selective adsorption of longer RNAs, at least for oligoadenylates on hydroxyapatite,10 consistent with its chromatographic properties.11 However, the idea that mineral surfaces might select for higher-mass RNAs in prebiotic conditions has not been pursued further, despite the fact that such a simple selective factor may have widely facilitated the appearance of genetic information on the early Earth.12,13 On the contrary, research since Orgel's original report10 has generally shown an inverse correlation between the maximum number of binding oligonucleotides and their length on various minerals, because the number of oligonucleotides that can occupy a fixed surface area becomes greater if each polymer is shorter.14 These studies, however, considered one oligonucleotide species at a time, and did not investigate a mixture of multiple different length molecules. Therefore, the role and generality of the selective enrichment of longer RNAs on mineral surfaces remains unclear.
Selection of longer RNA molecules on mineral surfaces, if satisfactorily understood, could refine the RNA world hypothesis in several ways. First, the length of RNAs synthesized abiotically or by catalytic RNAs (ribozymes) is typically biased towards shorter variants.15–17 Mineral surfaces could counter this bias and help maintain the expansion of genetic information and facilitate the advent of ribozymal activity. Second, shorter RNAs generally have faster replication kinetics and outcompete longer sequences, and hence genetic information would be easily lost from incipient self-replicating systems.18 Mineral surfaces could protect longer RNAs and allow their long-term propagation. Third, cooperation between RNA molecules has been considered a plausible mechanism for catalyzing complex reactions19,20 or collective reproduction.21,22 The specific accumulation of long informational RNAs could promote cooperative interactions if the mechanism helps exclude non-cooperating molecules. Most of these hypotheses, however, require combination of an RNA's catalytic ability with minerals. Although a few studies showed ribozymal activity in the presence of minerals, at least montmorillonite,8,23,24 such a synergistic effect of minerals and a ribozyme has not been demonstrated.
Following up on the observation of longer oligoadenylates accumulating on hydroxyapatite,10 here we investigated the generality of the enrichment of longer RNAs on mineral surfaces, using short random RNAs and a ribozyme. We first tested the selection among fully random 8-, 12-, 16-, 20-, and 24-mer RNAs, which model potentially available RNAs on the early Earth,13,15 on five kinds of mineral grains: two iron sulfides (pyrite and pyrrhotite; FeS2 and FexS, respectively), an iron oxide (magnetite; Fe3O4), a carbonate (calcite; CaCO3), and a phosphate mineral (hydroxyapatite; Ca5(PO4)3(OH)), whose identity and purity were confirmed by X-ray diffraction and scanning electron microscopy (Fig. S1, ESI†). These minerals are all thought to have been abundant throughout the early Earth.1,25 At the neutral pH (7.0), as tested here, it is expected that some of the minerals (at least pyrite and pyrrhotite) bear a net negative surface charge.26,27 RNA also carries a negative charge at this pH, but it can efficiently adsorb even onto negatively charged mineral surfaces with divalent cations as mediators.28,29
We co-incubated the mixture of RNAs with each mineral in the presence of Mg2+. After incubating with any of the minerals, the concentration of longer RNAs collected from the surfaces increased compared to the bulk solution, whereas the concentration of shorter RNAs decreased, indicating that minerals selectively accumulated longer RNAs (Fig. 1A). Although a small portion of RNAs of unknown lengths may have remained on minerals without being collected (Fig. S3A and B, ESI†), the enrichment of shorter RNAs in the supernatant confirms the enrichment of longer RNAs on the surfaces (Fig. 1B). The degree to which RNA sizes were shifted in supernatants with different minerals could be explained partly by the estimated percentage of adsorbed RNA, 30–40% for pyrite, pyrrhotite, and magnetite, and 60% for calcite and apatite (Fig. S3C, ESI†). The potential role of total adsorption was confirmed by using a higher concentration of RNAs with calcite or hydroxyapatite (13% estimated adsorption), which resulted in little enrichment of shorter RNAs in the supernatant, while displaying similar RNA distribution on the surfaces (Fig. S4, ESI†).
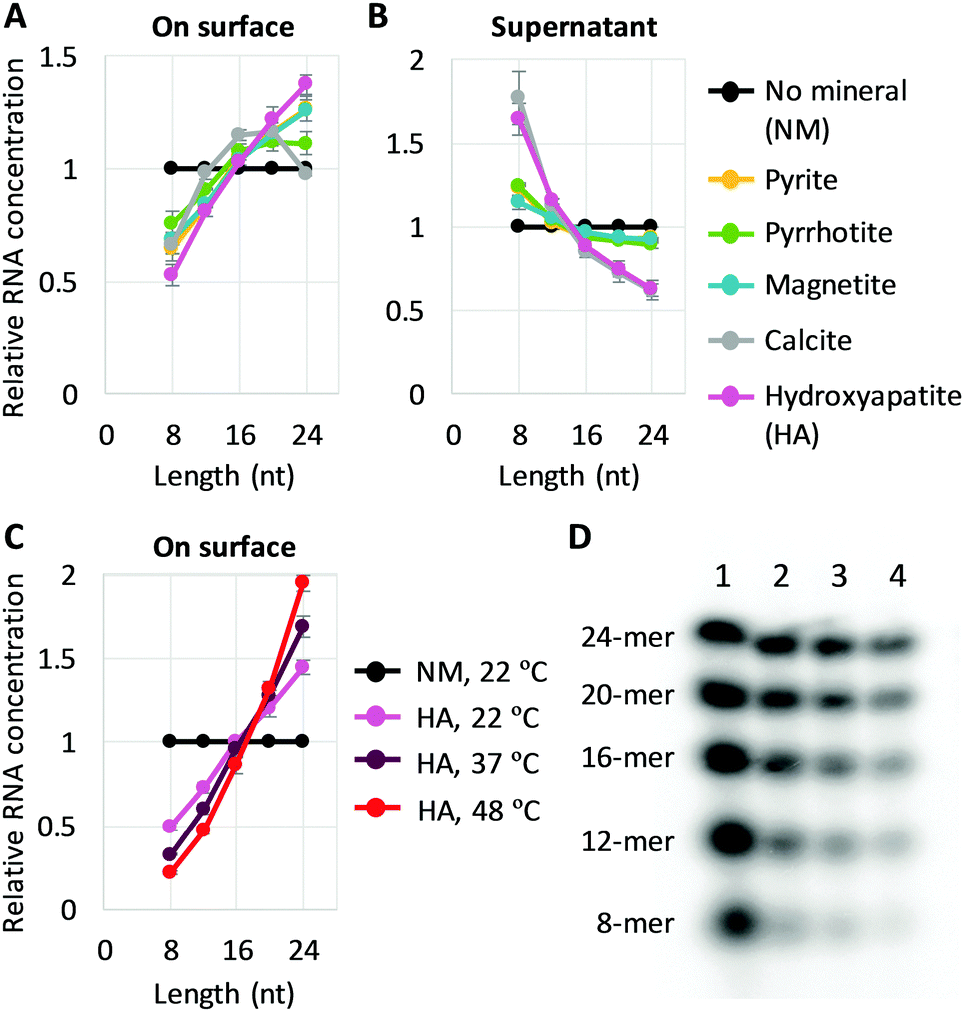 | ||
| Fig. 1 Longer RNA selection on mineral surfaces. (A and B) Random 8-, 12-, 16-, 20-, and 24-mer RNAs (0.6 μM each) and 0.2 mg of each mineral were incubated at 22 °C for 2 h, and the concentration of each length RNA on the surfaces (A) and in each supernatant (B) was determined from radioactivity of 32P-labeled RNAs through gel electrophoresis. An example of an analyzed gel image is shown in Fig. S2 (ESI†). (C) RNA concentration on the surface for adsorption experiments performed with hydroxyapatite (HA) at 22 °C, 37 °C, and 48 °C. (D) An example of a gel image. Lanes 1, no mineral (NM), 22 °C; 2, +HA, 22 °C; 3, +HA, 37 °C; 4, +HA, 48 °C. In all panels, RNA concentrations were normalized to the levels of a control reaction (NM) performed at 22 °C; the error bars indicate standard errors (N = 3). | ||
We also explored the sensitivity of length enrichment based on prebiotically relevant environmental parameters. We found that incubation at high temperatures increased the concentration of longer RNAs relative to shorter RNAs at least on pyrite, magnetite, and hydroxyapatite, in which hydroxyapatite showed the best enrichment (Fig. 1C, D and Fig. S5, ESI†). For example, the relative ratio of 24-mer RNAs to 8-mers on the hydroxyapatite surfaces increased from 2.9-fold to 8.7-fold with a temperature shift from 22 °C to 48 °C. These results indicate that high temperature could improve the relative enrichment of longer RNAs on mineral surfaces.
We then investigated whether the size-selection ability of mineral surfaces can be combined with catalytic reactions of a ribozyme. We used the Azoarcus self-assembly system (Fig. 2A; see Fig. S6, ESI† for more detail)19 for the catalytic reaction. In this system, the four fragments broken from the Azoarcus recombinase ribozyme, W, X, Y, and Z (39–63 nt), self-assemble to form a non-covalent complex that can catalyze covalent recombination of the fragments to create a series of elongated products (100–350 nt), including the ≈200 nt full-length ribozyme (WXYZ). We incubated the four fragments in the presence of hydroxyapatite and examined the distribution of products (Fig. 2B and C). We found that, on average, 19% of the total RNA was adsorbed (there was no uncollected RNA in a detectable level). Longer products were accumulated on mineral surfaces whereas no such tendency was observed in the supernatant. For example, ≈250 nt products (e.g., WXYZX) accumulated approximately 2.5-fold more than ≈100 nt products (WX) on the mineral surfaces. Notably, the longest 300–350 nt products accumulated to a detectable level only on the mineral surfaces. It should be noted that the reasonably short W fragment (63 nt) also accumulated more on the mineral surfaces relative to the supernatant (Fig. 2C), probably because of the formation of non-covalent WXYZ complexes.19 These results indicate that mineral surfaces can expand genetic complexity even when coupled with ribozyme function.
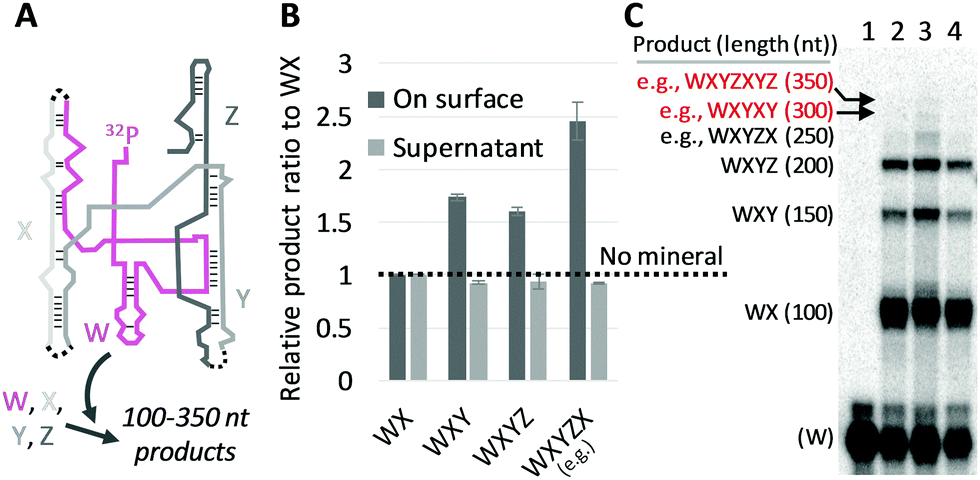 | ||
| Fig. 2 RNA adsorption on minerals coupled with Azoarcus self-assembling recombination. (A) Assembled Azoarcus ribozyme (WXYZ). The W fragment was partly 32P-labeled. Dotted lines show recombination junctions. (B) Ratio of some elongated products to WX, through the incubation of W, X, Y, and Z (2 μM each) at 48 °C for 2 h with 0.2 mg hydroxyapatite (HA). RNA concentrations (on surface, supernatant) were demonstrated by gel electrophoresis and normalized to the levels obtained with no minerals (NM, dotted line). The error bars indicate standard errors (N = 3). (C) An example of an analyzed gel image. Lanes 1, no reaction; 2, NM; 3, +HA, on surface; 4, +HA, supernatant; expected products and approximate lengths are indicated on the left. | ||
Next, to understand the underlying principle of the longer RNA selection on mineral surfaces, we constructed a simple mathematical model based on thermodynamics. Our derivation follows the approach for multisite adsorption from Ramirez-Pastor et al.30,31 and is detailed in the ESI.†
We started by considering a 1D lattice with M adsorption sites in contact with a large solution of each length (8-, 12-, 16-, 20- and 24-mer) RNA oligomer with a dimensionless concentration that remains constant at ![[c with combining macron]](https://www.rsc.org/images/entities/i_char_0063_0304.gif) = ck/c° (c° = 1 M). A k-mer occupies k sites upon adsorption, which is accompanied with a standard free energy change Δμads*, an affine function of length k:
= ck/c° (c° = 1 M). A k-mer occupies k sites upon adsorption, which is accompanied with a standard free energy change Δμads*, an affine function of length k: 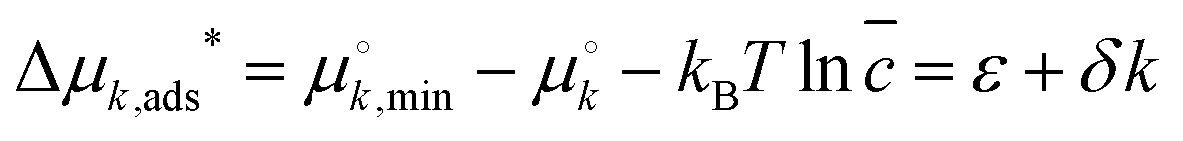 . Here,
. Here, 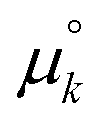 and
and 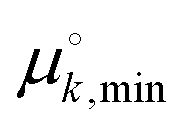 are standard free energies of formation for a k-mer in solution and on the mineral surface, respectively. kB is Boltzmann's constant and T is the absolute temperature. The parameter ε accounts for constant contributions to Δμk,ads*, such as the concentration term −kBT
are standard free energies of formation for a k-mer in solution and on the mineral surface, respectively. kB is Boltzmann's constant and T is the absolute temperature. The parameter ε accounts for constant contributions to Δμk,ads*, such as the concentration term −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln. The parameter δ accounts for contributions that scale with oligomer length k, such as molecular conformations and adsorption energy. Typically, ε > 0, because at micromolar concentration there is a large positive contribution by −kBT
ln. The parameter δ accounts for contributions that scale with oligomer length k, such as molecular conformations and adsorption energy. Typically, ε > 0, because at micromolar concentration there is a large positive contribution by −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln ≈ 14kBT. For adsorption to take place, a sufficient driving force needs to come from δ (mainly due to adsorption energy), implying δ < 0.
ln ≈ 14kBT. For adsorption to take place, a sufficient driving force needs to come from δ (mainly due to adsorption energy), implying δ < 0.
We also needed to consider the number of surface configurations (ways in which a mineral can be tiled by oligomers), denoted by Ω. In 1D, this simply corresponds to all unique ways in which we can place oligomers and empty sites in succession (Fig. 3A). The number of allowed surface configurations is thus a multinomial over the sites and oligomers. These mineral configurations provide an entropic contribution kBTlnΩ, which can be rewritten as
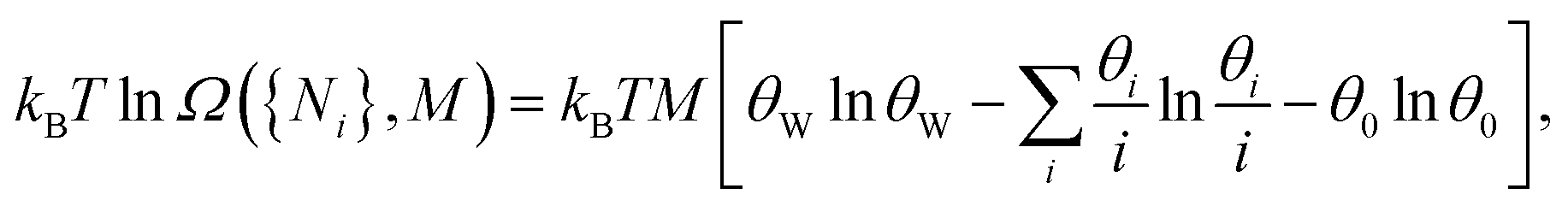 | (1) |
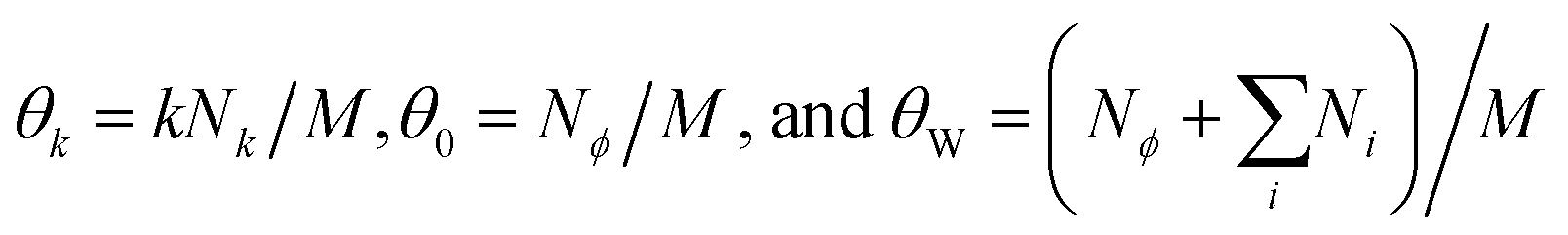 are the surface fraction occupied by a k-mer, the fraction of empty sites, and the fraction of empty sites and oligomers, respectively. The contribution is reminiscent of a mixing entropy, but with an extra θWlnθW term due to multisite adsorption. The 1D model can be extended to 2D surfaces with flexible polymers on the mineral (Fig. 3B).30,31 Surprisingly, these extensions amount to simply shifting the parameters ε and δ, meaning we retain the same simple model.
are the surface fraction occupied by a k-mer, the fraction of empty sites, and the fraction of empty sites and oligomers, respectively. The contribution is reminiscent of a mixing entropy, but with an extra θWlnθW term due to multisite adsorption. The 1D model can be extended to 2D surfaces with flexible polymers on the mineral (Fig. 3B).30,31 Surprisingly, these extensions amount to simply shifting the parameters ε and δ, meaning we retain the same simple model.
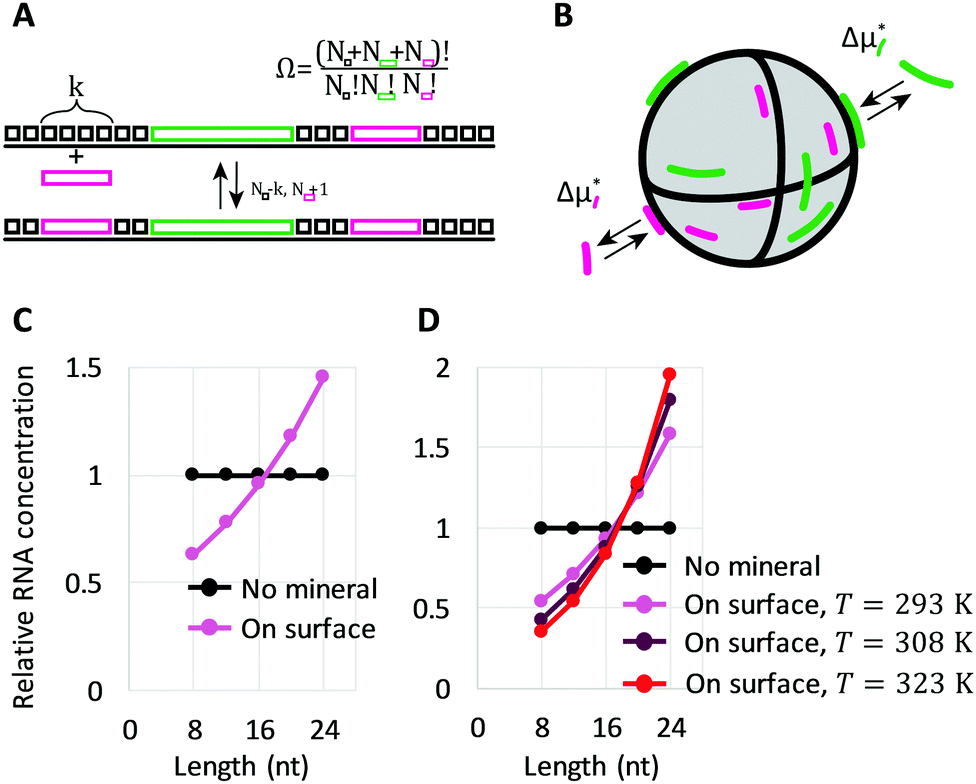 | ||
| Fig. 3 Longer RNA selection on mineral surfaces based on thermodynamics. (A) A reversible adsorption of a short oligomer to a 1D surface, covered with empty sites (black square boxes) and two different size oligomers (pink and green rectangles). Nk enumerates the number of each object. (B) Reversible adsorption of oligomers to mineral surfaces, associated with Δμk,ads*. (C) Relative oligomer concentrations on mineral surfaces at T = 293 K, following eqn (3) with ε(T*) = 3.5kBT*, δ(T*) = −0.5kBT*, and T* = 293 K. (D) Relative oligomer concentrations on mineral surfaces at three different temperatures T (293 K, 308 K, and 323 K), following eqn (3) with Δhε = −11.5kBT*, Δsε = 14.9kB, ε(T) = Δhε − TΔsε. Δhδ = −2.5kBT*, Δsδ = 1.7kB, and δ(T) = Δhδ − TΔsδ. The energy scale is set by reference temperature T* = 293 K. | ||
Putting the surface configurations and Δμk,ads* together, the surface coverage for a k-mer becomes
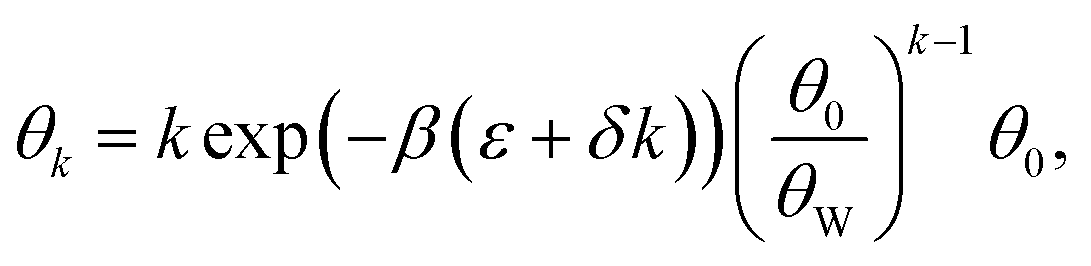 | (2) |
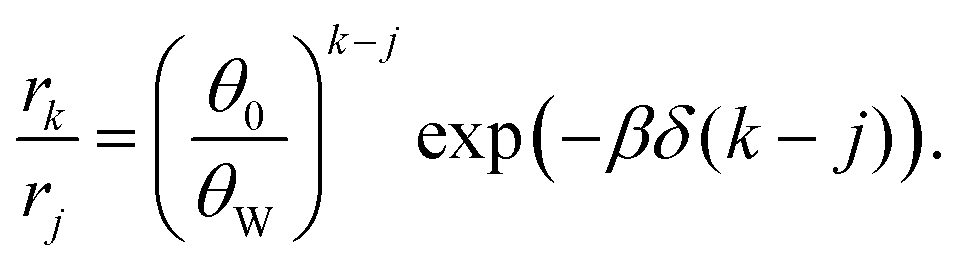 | (3) |
The factor (θ0/θW)k–j originates from surface configurations for mineral occupation. By definition, (θ0/θW) < 1, and thus it generally favors the adsorption of shorter oligomers. However, because δ < 0, the latter term favors the adsorption of longer oligomers. Fig. 3C shows an example of selection of longer RNA molecules. An instructive way to understand why longer oligomers can be favored is to imagine the contribution of the standard free energy change Δμ* for replacing an oligomer of length j by two oligomers of length j/2. Δμ* = 2Δμj/2* − Δμj* = ε. Since typically ε > 0, there is a penalty for covering the same surface with smaller oligomers, which becomes higher at low concentrations . Longer RNAs can therefore be thermodynamically selected on mineral surfaces (Fig. 3C), consistent with the experiment (Fig. 1A).
The free energy parameters ε and δ are composed of various entropic and enthalpic contributions (polymer configurations, interaction with solvents, interaction with minerals, etc.), which allows us to write: ε = Δhε − TΔsε and δ = Δhδ − TΔsδ. In order to inspect the temperature dependence, we therefore need two extra parameters (we will neglect any further temperature dependence of Δh and Δs). As shown in the ESI,† the derivative of eqn (3) with respect to temperature is proportional to
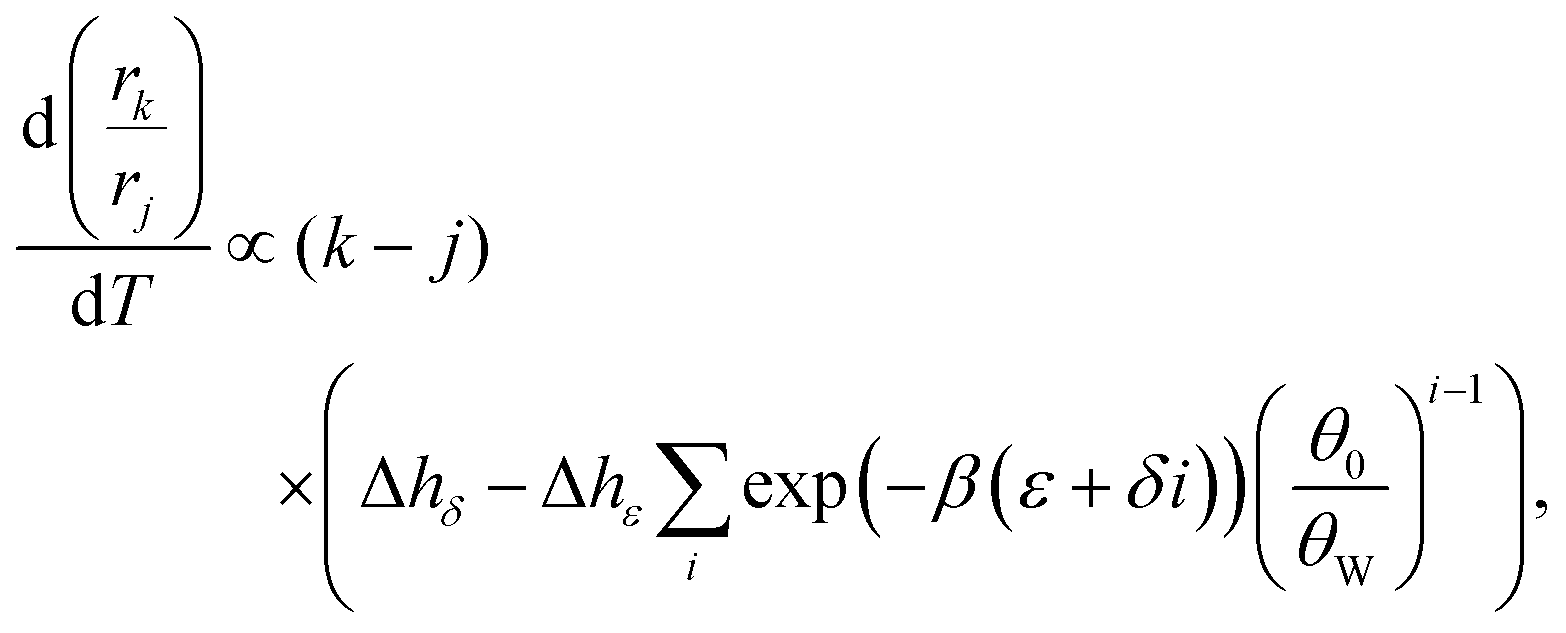 | (4) |
In this study, we demonstrated that a simple and ubiquitous environmental factor, mineral surfaces, can selectively enrich longer RNAs from a pool of short random RNA molecules. The tendency for size selection observed with a wide variety of minerals (Fig. 1A) suggests that the basis of this selection is not different chemical compositions of each mineral (e.g., different metals and charges) but surface structures, as confirmed by a mathematical model based on thermodynamic considerations (Fig. 3). Longer RNAs can be enriched more than shorter RNAs because of a general entropic cost of adsorption to surfaces. Moreover, not only did we find that a ribozyme can function with a prebiotic mineral other than montmorillonite,8,23,24 hydroxyapatite, we also showed that the recombinase ribozyme and the surfaces synergistically shifted the RNA length distribution upwards (Fig. 2B and C). These results support the plausibility that mineral surfaces selected for longer RNAs, since diverse minerals were distributed throughout the early Earth, including submarine sites and evaporitic field environments,1 both of which are candidate locales for the establishment of life.32–34 A recent study showed that thermophoresis and convection through porous environments, such as might occur at a deep-sea hydrothermal vent, could select longer oligonucleotides.35 Our study implies that the enrichment of longer genetic polymers may not have been restricted to specific milieus but could have occurred more widely on the early Earth. Slightly higher temperatures have been proposed for field environments,34 and these could have augmented length enrichment (Fig. 1C and 3D). It was previously shown that long RNAs adsorbed on mineral surfaces can promote RNA polymerization.36 Such a positive feedback could have helped increase genetic complexity and, thereby, facilitate the emergence of the first ribozymes, which could then cooperate with minerals to further develop genetic information (Fig. 2). Overall, our results expand the potential environments within which life-like processes could begin.
In conclusion, we provided experimental evidence and a theoretical explanation that mineral surfaces have an innate potential to selectively enrich longer RNA molecules and that such surfaces can cooperate with catalytic RNA to further increase genetic complexity. These results demonstrate that mineral surfaces, ubiquitous environments on the Earth, may have assisted in the expansion of genetic information in the origins of life.
We thank David Lacoste, Karyn Rogers, Ulysse Pedreira-Segade, and Vincent Riggi for useful discussion. This work was supported by funding from NASA to DB and NL (80NSSC17K0296) and Agence Nationale de la Recherche to AB (ANR-10-IDEX-0001-02, IRIS OCAV).
Conflicts of interest
There are no conflicts to declare.Notes and references
- H. J. Cleaves, A. M. Scott, F. C. Hill, J. Leszczynski, N. Sahai and R. Hazen, Chem. Soc. Rev., 2012, 41, 5502–5525 RSC.
- W. Gilbert, Nature, 1986, 319, 618 CrossRef.
- G. F. Joyce, Nature, 1989, 338, 217–224 CrossRef CAS PubMed.
- A. Ricardo, M. A. Carrigan, A. N. Olcott and S. A. Benner, Science, 2004, 303, 196 CrossRef CAS PubMed.
- G. Costanzo, R. Saladino, C. Crestini, F. Ciciriello and E. D. Mauro, J. Biol. Chem., 2007, 282, 16729–16735 CrossRef CAS PubMed.
- J. P. Ferris, A. R. Hill, R. Liu and L. E. Orgel, Nature, 1996, 381, 59–61 CrossRef CAS PubMed.
- J. P. Ferris, Philos. Trans. R. Soc., B, 2006, 361, 1777–1786 CrossRef CAS PubMed.
- E. Biondi, S. Branciamore, M. C. Maurel and E. Gallori, BMC Evol. Biol., 2007, 7, 1–7 CrossRef PubMed.
- E. Biondi, Y. Furukawa, J. Kawai and S. A. Benner, Beilstein J. Org. Chem., 2017, 13, 393–404 CrossRef CAS PubMed.
- D. Gibbs, R. Lohrmann and L. E. Orgel, J. Mol. Evol., 1980, 15, 347–354 CrossRef CAS PubMed.
- G. Bernardi, Nature, 1965, 206, 779–783 CrossRef CAS PubMed.
- G. Wächtershäuser, Microbiol. Rev., 1988, 52, 452–484 Search PubMed.
- C. Briones, M. Stich and S. C. Manrubia, RNA, 2009, 15, 743–749 CrossRef CAS PubMed.
- H. J. Cleaves, E. Crapster-Pregont, C. M. Jonsson, C. L. Jonsson, D. A. Sverjensky and R. A. Hazen, Chemosphere, 2011, 83, 1560–1567 CrossRef PubMed.
- P. A. Monnard, A. Kanavarioti and D. W. Deamer, J. Am. Chem. Soc., 2003, 125, 13734–13740 CrossRef CAS PubMed.
- J. Attwater, A. Wochner and P. Holliger, Nat. Chem., 2013, 5, 1011–1018 CrossRef CAS PubMed.
- C. Gibard, S. Bhowmik, M. Karki, E. Kim and R. Krishnamurthy, Nat. Chem., 2017, 10, 212–217 CrossRef PubMed.
- D. R. Mills, R. L. Peterson and S. Spiegelman, Proc. Natl. Acad. Sci. U. S. A., 1967, 58, 217–224 CrossRef CAS.
- E. J. Hayden and N. Lehman, Chem. Biol., 2006, 13, 909–918 CrossRef CAS PubMed.
- H. Mutschler, A. Wochner and P. Holliger, Nat. Chem., 2015, 7, 502–508 CrossRef CAS PubMed.
- M. Eigen and P. Schuster, Naturwissenschaften, 1977, 64, 541–565 CrossRef CAS PubMed.
- N. Vaidya, M. L. Manapat, I. A. Chen, R. Xulvi-Brunet, E. J. Hayden and N. Lehman, Nature, 2012, 491, 72–77 CrossRef CAS PubMed.
- E. Biondi, S. Branciamore, L. Fusi, S. Gago and E. Gallori, Gene, 2007, 389, 10–18 CrossRef CAS PubMed.
- J. D. Stephenson, M. Popović, T. F. Bristow and M. A. Ditzler, RNA, 2016, 22, 1893–1901 CrossRef CAS PubMed.
- R. M. Hazen and J. M. Ferry, Elements, 2010, 6, 9–12 CrossRef CAS.
- M. J. Dekkers and M. A. A. Schoonen, Geochim. Cosmochim. Acta, 1994, 58, 4147–4153 CrossRef CAS.
- J. Bebié and M. A. A. Schoonen, Geochem. Trans., 2000, 1, 47–53 CrossRef.
- M. Franchi, J. P. Ferris and E. Gallori, Origins Life Evol. Biospheres, 2003, 33, 1–16 CrossRef CAS.
- J. A. Libera, H. Cheng, M. Olvera, D. Cruz and M. J. Bedzyk, J. Phys. Chem. B, 2005, 109, 23001–23007 CrossRef CAS PubMed.
- A. J. Ramirez-Pastor, T. P. Eggarter, V. D. Pereyra and J. L. Riccardo, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 11027–11036 CrossRef CAS.
- F. Romá, J. L. Riccardo and A. J. Ramirez-Pastor, Ind. Eng. Chem. Res., 2006, 45, 2046–2053 CrossRef.
- W. Martin, J. Baross, D. Kelley and M. J. Russell, Nat. Rev. Microbiol., 2008, 6, 805–814 CrossRef CAS PubMed.
- B. Damer and D. Deamer, Life, 2015, 5, 872–887 CrossRef CAS PubMed.
- B. K. D. Pearce, R. E. Pudritz, D. A. Semenov and T. K. Henning, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11327–11332 CrossRef CAS PubMed.
- M. Kreysing, L. Keil, S. Lanzmich and D. Braun, Nat. Chem., 2015, 7, 203–208 CrossRef CAS PubMed.
- O. L. Acevedo and L. E. Orgel, Nature, 1986, 321, 790–792 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, full details of mathematical models, Fig. S1–S7. See DOI: 10.1039/c8cc10319d |
| This journal is © The Royal Society of Chemistry 2019 |