
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2,7-Diazabicyclo[2.2.1]heptanes: novel asymmetric access and controlled bridge-opening†
Gary R.
Peczkowski
a,
Philip G. E.
Craven
 a,
Darren
Stead
b and
Nigel S.
Simpkins
*a
a,
Darren
Stead
b and
Nigel S.
Simpkins
*a
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: N.Simpkins@bham.ac.uk
bMedicinal Chemistry, Oncology, IMED Biotech Unit, AstraZeneca, Cambridge, CB4 0WG, UK
First published on 12th March 2019
Abstract
Organocatalysed asymmetric Michael additions of substituted triketopiperazines to enones afford products in high yield and enantiomeric ratio (er). Further modification delivers products possessing natural product (NP) scaffolds including diazabicyclo[2.2.1]heptane, prolinamide and harmicine.
For many years natural products have provided organic chemists with a multitude of synthetically challenging architectures, providing the stimulus for the development of new synthetic methods and strategies. An additional driving force in this field is provided by the diverse biological activities inherent in many natural products. The fact that around 30% of small molecule drugs developed in the last 30 years are either natural products, or their close relatives, is ample testament to the significance of this area.1,2
Traditionally, natural product synthesis is the exploration of a route towards a singular target compound3 and with rapid access in mind, many research groups compete for the shortest linear sequence.4,5 However, this approach often overlooks any potentially rewarding analogues or family members.6 Therefore, a route in which multiple interesting scaffolds may be accessed from a common motif is highly desirable.7,8
In this report we describe how our newly developed asymmetric access to triketopiperazines (TKPs) can be utilised in this way to gain access to several interesting natural product-like motifs, as illustrated in Scheme 1.
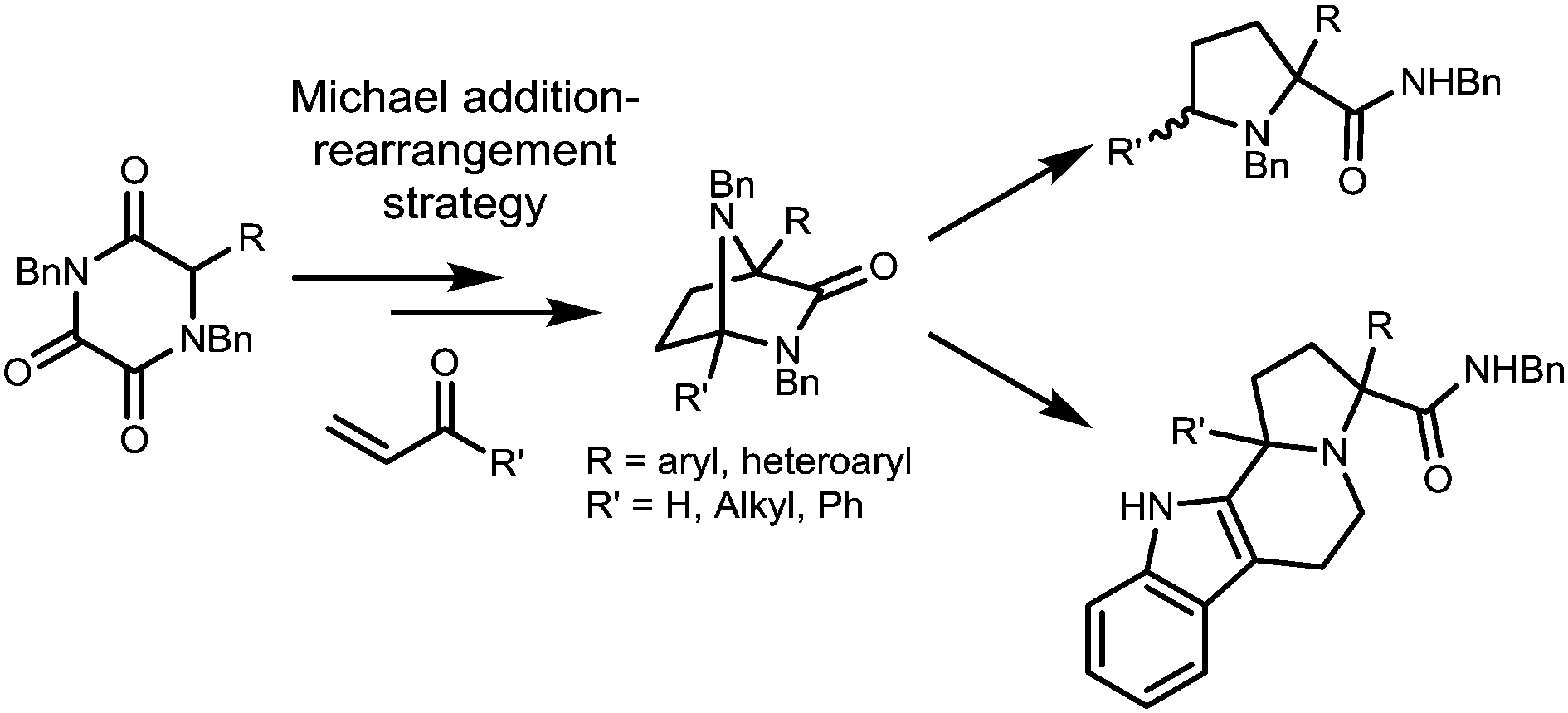 | ||
| Scheme 1 Michael addition-rearrangement strategy towards NP scaffolds. | ||
We conceived that structures including various diazabicyclo[2.2.1]heptane core structures and proline amides would be available via initial asymmetric Michael addition of a TKP, followed by TKP manipulations involving ring-opening. The diazabicyclo[2.2.1]heptane structures are especially interesting as core components of biologically active products as shown in Fig. 1.9–15
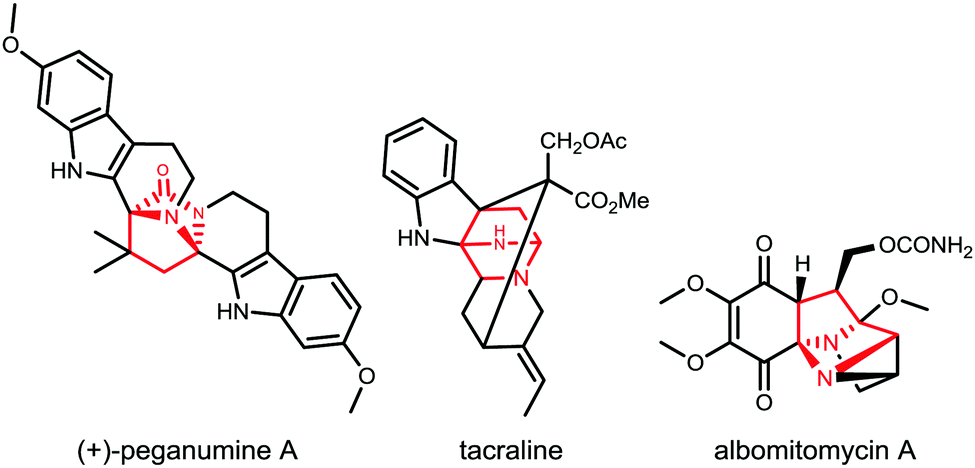 | ||
| Fig. 1 NPs containing the diazabicyclo[2.2.1]heptane motif. | ||
Recent reports from our group have described highly enantioselective Michael addition chemistry of various substituted TKP systems.16–18 The chiral TKPs are generated in highly diastereo- and enantioselective form, and are amenable to fruitful regioselective manipulation to give attractive natural product like structures.
In the present research we opted to employ novel TKP structures in which R = α-aryl and α-heteroaryl, in order to achieve the objectives shown in Scheme 1. Initially, α-aryl TKPs 1a–j were readily prepared using previously established methods.16,18–24 All optimisation was conducted with phenyl TKP 1a employing methyl vinyl ketone (MVK) as the Michael acceptor (for details see ESI†).25 Under the optimised conditions, using the O-benzylated cinchona derived catalyst 3, the Michael additions of α-aryl TKPs 1a–j to MVK were explored and the results are shown in Table 1 (entries 1–10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | TKP | Ar | R | 2 (%) | er |
| a −30 °C, 12 days. b HPLC conducted on the acetal derivative. | |||||
| 1 | 1a | Ph | Me | 2a 90 | 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 :8 |
| 2 | 1b | p-C6H4OMe | Me | 2b 83 | 93:7 |
| 3 | 1c | p-C6H4NO2 | Me | 2c 70 | 95:5 |
| 4 | 1d | p-C6H4Br | Me | 2d 88 | 91:9 |
| 5 | 1e | Furan-2-yl | Me | 2e 99 | 94:6 |
| 6 | 1f | Thiophen-2-yl | Me | 2f 93 | 94:6 |
| 7 | 1g | o-C6H4Br | Me | 2g 88 | 55:44 |
| 8 | 1h | N-Methylpyrrol-2-yl | Me | 2h 99 | 50:50 |
| 9 | 1i | N-Methylpyrrol-3-yl | Me | 2i 63 | 77:23 |
| 10 | 1j | Indol-3-yl | Me | 2j 91 | 73:27 |
| 11 | 1a | Ph | Et | 2k 91 | 96:4 |
| 12 | 1b | p-C6H4OMe | Et | 2l 75 | 97:3 |
| 13 | 1c | p-C6H4NO2 | Et | 2m 63 | 97:3 |
| 14 | 1a | Ph | Ph | 2n 90 | 85:15 |
| 15 | 1b | p-C6H4OMe | Ph | 2o 95 | 87:13 |
| 16 | 1c | p-C6H4NO2 | Ph | 2p 88 | 96:4 |
| 17 | 1a | Ph | H | 2q 85 | 58:42a,b |
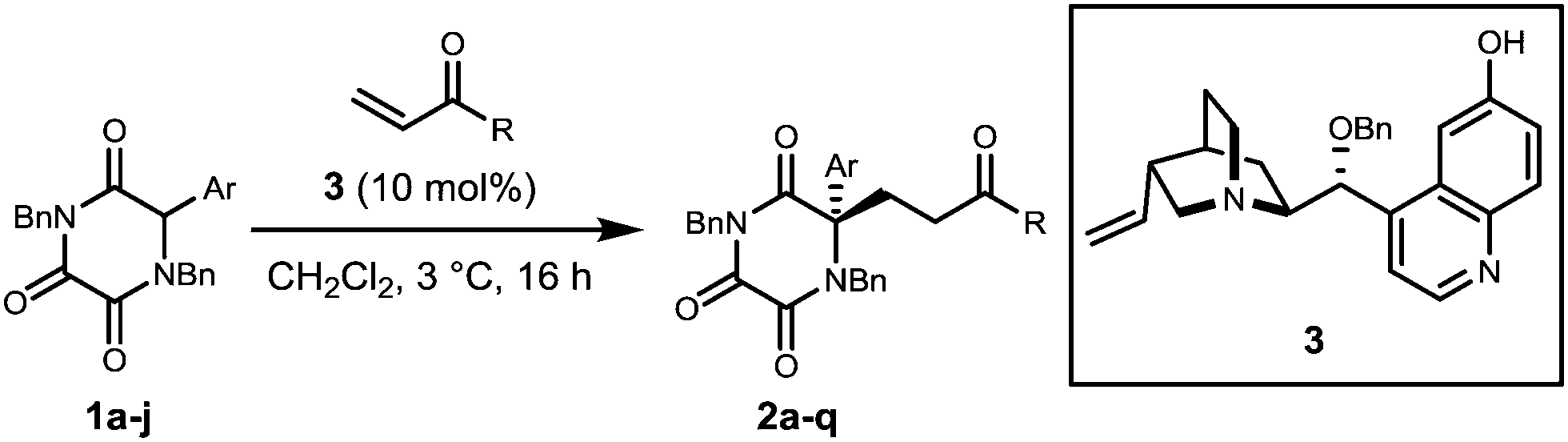
Addition products 2a–j were isolated in good to excellent yields albeit with mixed levels of enantioselectivity. High levels of enantioselectivity were observed for substrates possessing either phenyl, p-substituted phenyl or unsubstituted 5-membered heterocycles (entries 1–6). Erosion of enantioselectivity appears to be the result of increased steric requirement proximal to the reacting enolate centre, e.g. with o-substituted aryl substrates (entries 7 and 8). This is exemplified by two N-Me pyrrole analogues (entries 8 and 9) in which moving the N-methyl group away from the reactive centre results in a significant increase in enantioselectivity.
When these conditions were employed with alternative enone acceptors, varied results were obtained (entries 11–17). Enantioselectivities remained high when employing ethyl vinyl ketone (entries 11–13) while a marked reduction in selectivity was observed with the use of phenyl vinyl ketone (entries 14 and 15) with the exception of entry 16 at 96:4 er. Selectivity was vastly reduced when using acrolein as the Michael acceptor, even when the reaction was performed at lower temperature (entry 17).
Following crystallisation, the absolute configuration of adduct 2f, generated from reaction of TKP 1f with MVK, was determined by X-ray crystallography (Fig. 2A).26
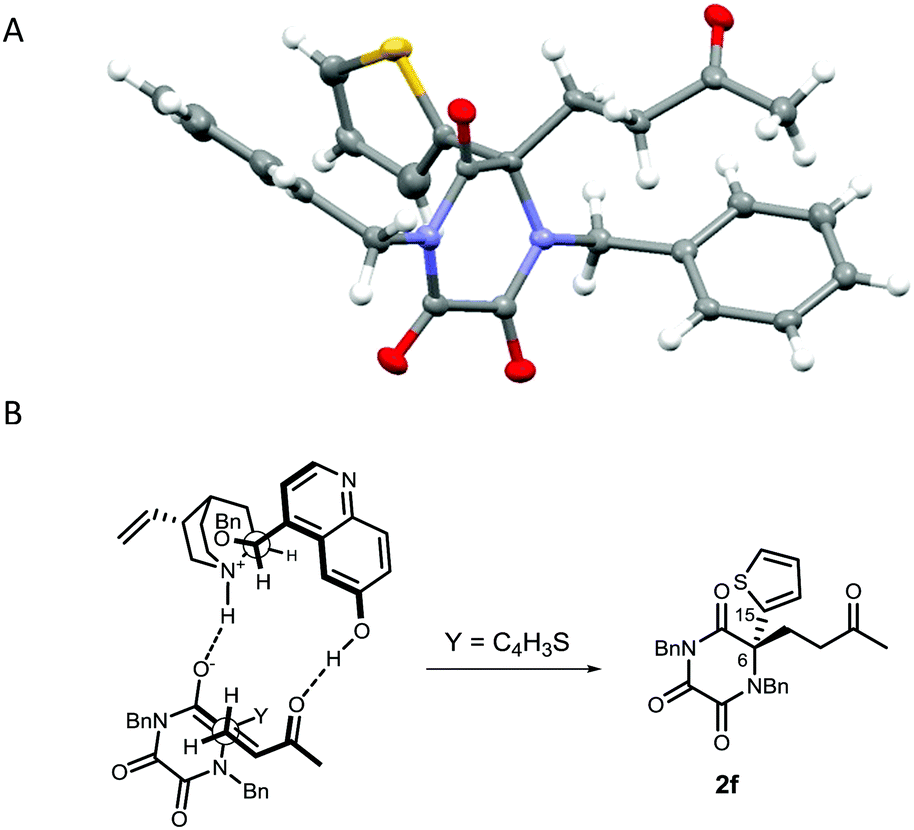 | ||
| Fig. 2 (A) Crystal structure of 2f with ellipsoids drawn at the 50% probability level. The thiophene ring is disordered over two positions, which is the result of rotation around the C6–C15 bond, at a refined percentage occupancy ratio of 63.9(3):36.1(3), only the major position is shown for clarity. (B) Model for 3-catalysed Michael addition generating 2f. | ||
Our results are in accordance with the stereochemical model originally proposed by Deng for the dual activation and co-ordination, by the catalyst, of both the nucleophile and the electrophile, and with our group's previous works (Fig. 2B).16,17,27
With the Michael addition products 2 in hand, our attention turned to the removal of the oxalyl unit in an attempt to free the quaternary α-amino acid. It was envisioned that the use of a dinucleophile would achieve this goal through initial attack at the highly electrophilic C-3 carbonyl and subsequent cyclisation onto the neighbouring carbonyl.16 After investigating a range of dinucleophiles, it was found that ethanolamine removed the oxalyl component, however, the free aminoamide was not isolated. Instead, direct access to products that possess the diazabicyclo[2.2.1]heptane (4) core framework was achieved, Table 2.
|
|
||||
|---|---|---|---|---|
| Entry | TKP | Ar | R | 4 (%) |
| a Enantioenriched substrates retain enantiopurity. | ||||
| 1 | 2a | Ph | Me | 4a 87a |
| 2 | 2b | p-C6H4OMe | Me | 4b 51 |
| 3 | 2c | p-C6H4NO2 | Me | 4c 61 |
| 4 | 2d | p-C6H4Br | Me | 4d 60 |
| 5 | 2e | Furan-2-yl | Me | 4e 75 |
| 6 | 2f | Thiophen-2-yl | Me | 4f 50a |
| 7 | 2g | o-C6H4Br | Me | 4g 29 |
| 8 | 2h | N-Methylpyrrol-2-yl | Me | 4h 26 |
| 9 | 2i | N-Methylpyrrol-3-yl | Me | 4i 25 |
| 10 | 2j | Indol-3-yl | Me | 4j 21 |
| 11 | 2k | Ph | Et | 4k 50 |
| 12 | 2n | Ph | Ph | 4l 84 |
| 13 | 2q | Ph | H | 4m 28 |
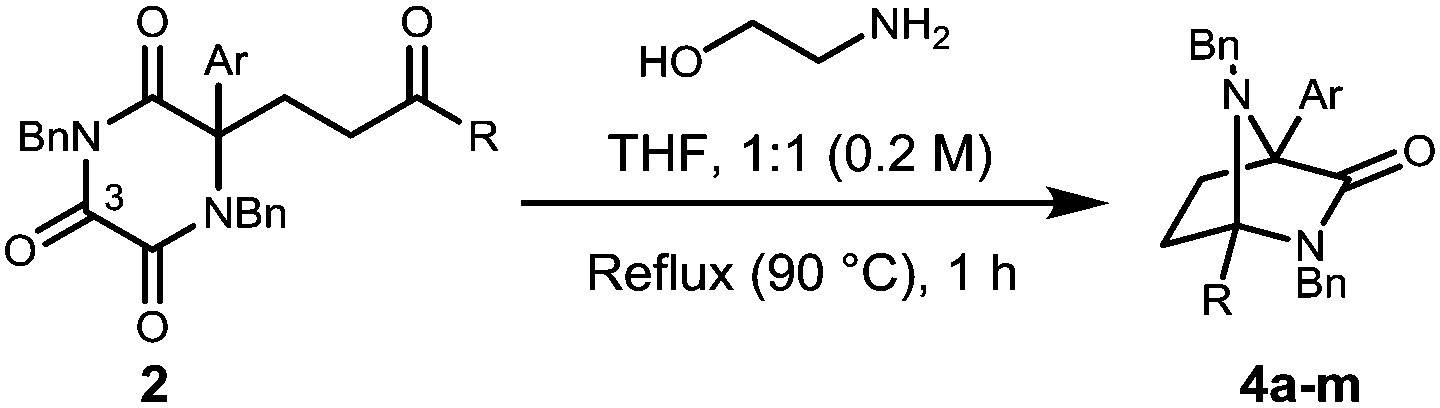
This process involves release of the TKP ring nitrogen functions from the oxalyl unit, and their convergent condensation with the ketone originating from the Michael acceptor to form an unusual bridged N-acyl aminal structure. Moderate to high yields were observed in most cases (entries 1–6, 11 and 12) with the exception of substrates bearing o-substituents (entries 7–10) and the acrolein adduct (entry 13). As expected, the conversion of enantioenriched substrates proceeded with retention of enantiopurity, as demonstrated for entries 1 and 6. The structure of 4a was unequivocally confirmed by single crystal X-ray analysis (Fig. 3).28
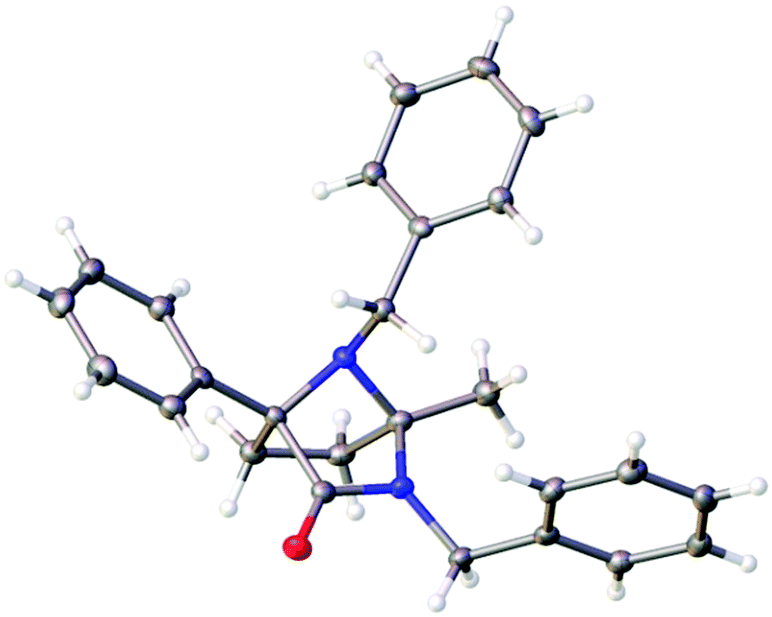 | ||
| Fig. 3 Crystal structure of 4a with ellipsoids drawn at the 50% probability level. | ||
With diazabicycle 4a in hand, we endeavoured to break open the newly formed cyclic aminal to reveal a quaternary 2,5-disubstituted prolinamide. Several previously described reductive ring opening conditions were examined and details can be found in the ESI.†29–31
Reduction of diazabicycle 4a with DIBAL at −78 °C affords prolinamides 5a and 5b in a modest 6.5:1 dr with a combined yield of 57% (Scheme 2).
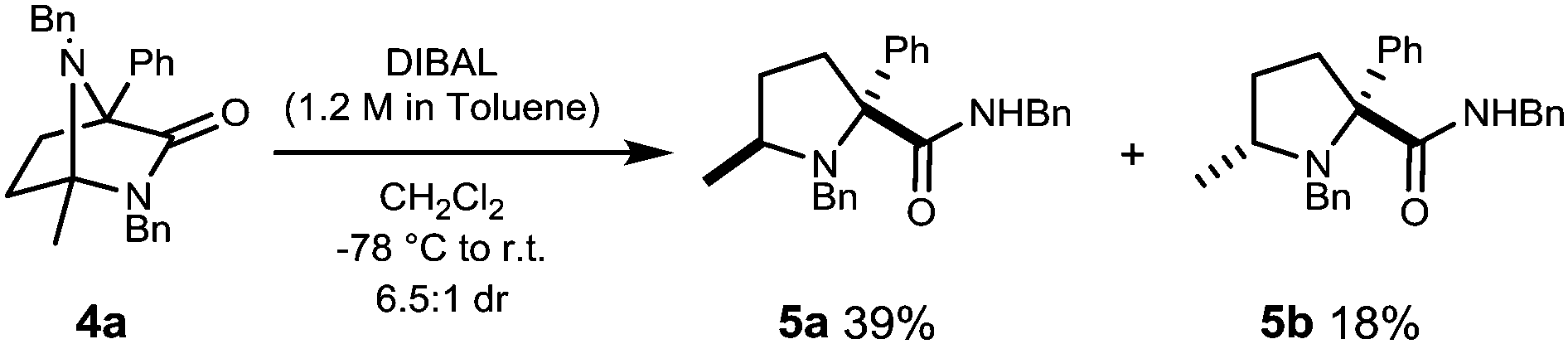 | ||
| Scheme 2 Reductive ring opening of 4a. | ||
Interestingly, the putative intermediate iminium ion involved in the process can be isolated in the form of 6 by treatment of diazabicycle 4a with HCl in dioxane. Preliminary investigations have found that this also undergoes reduction to give prolinamides 5a + b (Scheme 3).
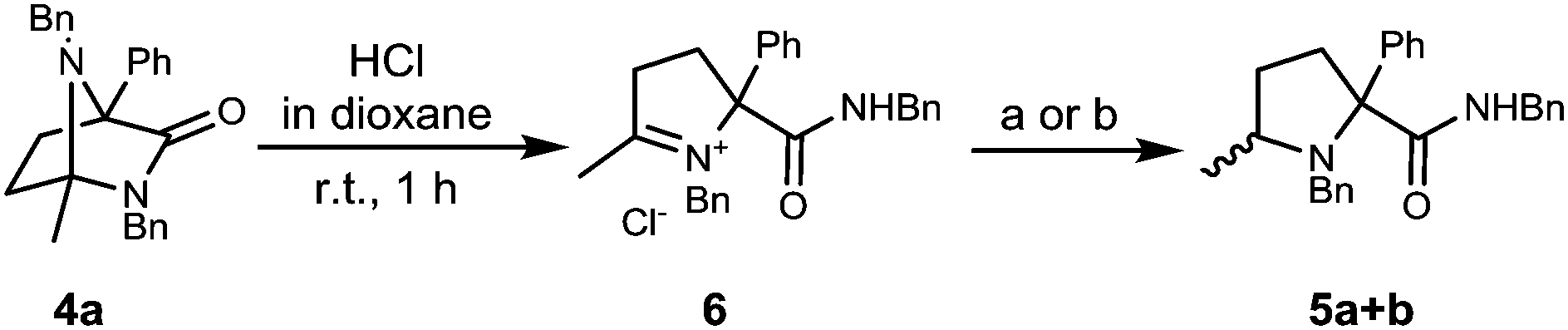 | ||
| Scheme 3 Reaction pathway to prolinamides 5a + b (a) L-selectride, 1.0:1.3 dr; (b) LiAlH4, 1.5:1.0 dr. | ||
The potential to access further natural product skeletons was investigated by intramolecular trapping of the iminium intermediate. Thus, diazabicycle 9 was synthesised from TKP 7 using our optimised methodology (Scheme 4). Treatment of 9 with HCl instigated a Pictet–Spengler reaction to yield product 10, as a single isomer, which possesses the scaffold of the natural product harmicine.32,33
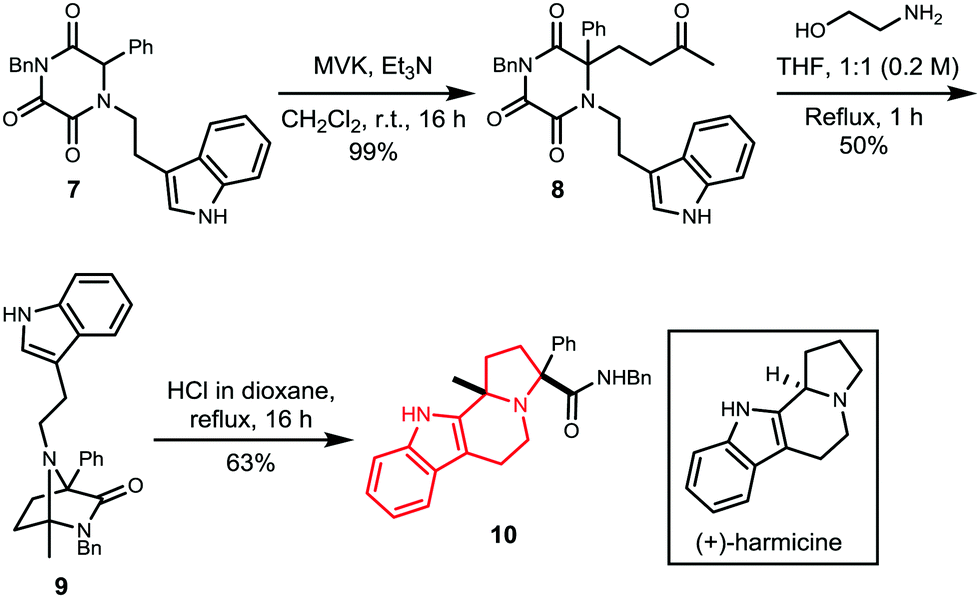 | ||
| Scheme 4 Synthesis of harmicine derivative 10. | ||
In conclusion, we have demonstrated that α-aryl TKPs can undergo asymmetric Michael additions with high selectivity and that manipulation of the TKP motif allows access to multiple products possessing NP scaffolds. We anticipate that additional biologically relevant and NP scaffolds can be accessed and investigations are currently on-going within our laboratory to unlock the further potential of the TKP motif.
We acknowledge the University of Birmingham, EPSRC and AstraZeneca for support of GP. The NMR and HPLC instruments used in this research were obtained through Birmingham Science City: Innovative uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). We also thank Dr Louise Male, Dr Cécile Le Duff, Dr Chi Tsang, Dr Peter Ashton and Dr Allen Bowden and the Centre for Chemical and Materials Analysis in the School of Chemistry at the University of Birmingham for analytical support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef CAS PubMed.
- A. L. Harvey, Drug Discovery Today, 2008, 13, 894–901 CrossRef CAS PubMed.
- F. Saint-Dizier and N. S. Simpkins, Chem. Sci., 2017, 8, 3384–3389 RSC.
- J. W. Reed and T. Hudlicky, Acc. Chem. Res., 2015, 48, 674–687 CrossRef CAS PubMed.
- P. S. Baran, T. J. Maimone and J. M. Richter, Nature, 2007, 446, 404–408 CrossRef CAS PubMed.
- J. Shimokawa, Tetrahedron Lett., 2014, 55, 6156–6162 CrossRef CAS.
- S. M. Geddis, L. Carro, J. T. Hodgkinson and D. R. Spring, Eur. J. Org. Chem., 2016, 5799–5802 CrossRef CAS PubMed.
- J. T. Njardarson, C. Gaul, D. Shan, X. Y. Huang and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 1038–1040 CrossRef CAS PubMed.
- M. Kono, Y. Saitoh, K. Shirahata, Y. Arai and S. Ishii, J. Am. Chem. Soc., 1987, 109, 7224–7225 CrossRef CAS.
- J. E. Saxton, Nat. Prod. Rep., 1997, 14, 559 RSC.
- C. Piemontesi, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2016, 138, 11148–11151 CrossRef CAS PubMed.
- K. B. Wang, Y. T. Di, Y. Bao, C. M. Yuan, G. Chen, D. H. Li, J. Bai, H. P. He, X. J. Hao, Y. H. Pei, Y. K. Jing, Z. L. Li and H. M. Hua, Org. Lett., 2014, 16, 4028–4031 CrossRef CAS PubMed.
- T. A. Van Beek, R. Verpoorte, A. B. Svendsen and R. Fokkens, J. Nat. Prod., 1985, 48, 400–423 CrossRef CAS.
- S. R. Rajski and R. M. Williams, Chem. Rev., 1998, 98, 2723–2796 CrossRef CAS PubMed.
- P. D. Bass, D. A. Gubler, T. C. Judd and R. M. Williams, Chem. Rev., 2013, 113, 6816–6863 CrossRef CAS PubMed.
- A. Cabanillas, C. D. Davies, L. Male and N. S. Simpkins, Chem. Sci., 2015, 6, 1350–1354 RSC.
- M. Rees, N. S. Simpkins and L. Male, Org. Lett., 2017, 19, 1338–1341 CrossRef CAS PubMed.
- R. W. Foster, E. N. Lenz, N. S. Simpkins and D. Stead, Chem. – Eur. J., 2017, 23, 8810–8813 CrossRef CAS PubMed.
- A. R. Katritzky, A. T. Cutler, N. Dennis, G. J. Sabongi, S. Rahimi-Rastgoo, G. W. Fischer and I. J. Fletcher, J. Chem. Soc., Perkin Trans. 1, 1980, 1176–1184 RSC.
- K. A. Mix and R. T. Raines, Org. Lett., 2015, 17, 2358–2361 CrossRef CAS PubMed.
- C. R. Whitlock and M. P. Cava, Tetrahedron Lett., 1994, 35, 371–374 CrossRef CAS.
- S. Crooke and C. Whitlock, Molecules, 2012, 17, 14841–14845 CrossRef CAS PubMed.
- N. A. Petasis, A. Goodman and I. A. Zavialov, Tetrahedron, 1997, 53, 16463–16470 CrossRef CAS.
- S. R. K. Pingali, S. K. Upadhyay and B. S. Jursic, Green Chem., 2011, 13, 928–933 RSC.
- ESI†.
- The CIF for the crystal structure of 2f has been deposited with the CCDC and given the deposition number, CCDC 1880502†.
- H. Li, Y. Wang, L. Tang, F. Wu, X. Liu, C. Guo, B. M. Foxman and L. Deng, Angew. Chem., Int. Ed., 2005, 44, 105–108 CrossRef CAS PubMed.
- The CIF for the crystal structure of 4a has been deposited with the CCDC and given the deposition number, CCDC 1880503†.
- M. Sannigrahi, P. Pinto, T. M. Chan, N. Y. Shih and F. George Njoroge, Tetrahedron Lett., 2006, 47, 4877–4880 CrossRef CAS.
- M. Kasai, M. Kono, S. Ikeda, N. Yoda and N. Hirayama, J. Org. Chem., 1992, 57, 7296–7299 CrossRef CAS.
- Y. Zhang, L. Zheng, F. Yang, Z. Zhang, Q. Dang and X. Bai, Tetrahedron, 2015, 71, 1930–1939 CrossRef CAS.
- I. T. Raheem, P. S. Thiara, E. A. Peterson and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 13404–13405 CrossRef CAS PubMed.
- T.-S. Kam and K.-M. Sim, Phytochemistry, 1998, 47, 145–147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, NMR spectra and X-ray data. CCDC 1880502 and 1880503. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc10263e |
| This journal is © The Royal Society of Chemistry 2019 |