
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA new strategy for the in vitro selection of stapled peptide inhibitors by mRNA display†
Emil S.
Iqbal
 ab,
Stacie L.
Richardson
ab,
Nicolas A.
Abrigo
ab,
Kara K.
Dods
ab,
H. Estheban
Osorio Franco
ab,
Heather S.
Gerrish
ab,
Hari Kiran
Kotapati
c,
Iain M.
Morgan
bd,
Douglas S.
Masterson
c and
Matthew C. T.
Hartman
*ab
ab,
Stacie L.
Richardson
ab,
Nicolas A.
Abrigo
ab,
Kara K.
Dods
ab,
H. Estheban
Osorio Franco
ab,
Heather S.
Gerrish
ab,
Hari Kiran
Kotapati
c,
Iain M.
Morgan
bd,
Douglas S.
Masterson
c and
Matthew C. T.
Hartman
*ab
aDepartment of Chemistry, Virginia Commonwealth University, 1001 West Main Street, P. O. Box 842006, Richmond, VA 23284, USA. E-mail: mchartman@vcu.edu
bMassey Cancer Center, Virginia Commonwealth University, 401 College Street, Richmond, VA 23298, USA
cSchool of Mathematics & Natural Sciences, Chemistry & Biochemistry, 118 College Drive #5043, Hattiesburg, MS 39406, USA
dPhilips Institute for Oral Health Research, Department of Oral and Craniofacial Molecular Biology, School of Dentistry, Virginia Commonwealth University, Box 980566, Richmond, VA 23298, USA
First published on 10th July 2019
Abstract
Hydrocarbon stapled peptides are promising therapeutics for inhibition of intracellular protein–protein interactions. Here we develop a new high-throughput strategy for hydrocarbon stapled peptide discovery based on mRNA display of peptides containing α-methyl cysteine and cyclized with m-dibromoxylene. We focus on development of a peptide binder to the HPV16 E2 protein.
Approximately 80% of the proteome is non-enzymatic and intracellular, which presents a considerable challenge to current therapeutic development. Overall, small molecule drugs as a class have had limited success as inhibitors of protein–protein interactions (PPIs).1,2 Thousands of these interactions (over 60% of the structures in the protein data bank) involve an alpha-helix at the interface.3,4 Hydrocarbon stapled peptides have emerged as promising therapeutic strategies for targeting these interactions, even entering clinical trials.5 Hydrocarbon stapling involves replacing two residues on a single face of the helix with olefin containing amino acids followed by cyclization by olefin metathesis.6 While a promising strategy, the route from the peptide sequence at the interface to the final high-affinity hydrocarbon stapled peptide hit requires significant and laborious optimization (e.g.ref. 7 and 8). Moreover, in some cases, the linear peptides excised from the interface have low inherent affinity that cannot be rescued through stapling and subsequent rational design.9 A high-throughput approach to hydrocarbon staple discovery and optimization offers a potential solution to these problems.
There have been a few efforts to create libraries of stapled peptides, but so far the diversities of these libraries have been limited to around 100 unique peptides.10,11 It would be very exciting to use in vitro selection strategies (like phage display, mRNA display) to create stapled peptide libraries, as these libraries afford the ability to create peptide libraries that contain up to 1013 members. Moreover, mRNA display allows for the direct incorporation of non-canonical amino acids (ncAAs) into peptide libraries for further optimization and enhancement of affinity.12–14 But the olefin metathesis chemistry used for cyclization of the staple is incompatible with the aqueous conditions and the numerous nucleophiles present in these display systems. To provide bio-compatible alternatives for stapling, there has been a recent explosion in staple chemistries focused on cysteine alkylation.15 Some of these staple mimetics endow peptides with helical character16–18 and cell permeability.19,20 Heinis, for example, used cysteine alkylation in order find high affinity helical binders to β-catenin21 using a diverse phage displayed library.
But one aspect that each of these new cysteine stapling chemistries has neglected is the presence of the alpha-methyl substituent in standard hydrocarbon stapled peptides. The alpha-methyl substituent is not always required for retaining the binding affinity of hydrocarbon stapled peptides,22 but it does improve helicity and increase hydrophobicity—both of these aspects can enhance the cellular activity of these peptides.18,23,24 In this paper we investigate the properties of cyclic peptides containing alpha-methyl Cys (α-MeCys), highlighting the importance of the alpha-methyl group for hydrocarbon staple mimicry, and develop a system for incorporation of α-MeCys into diverse peptide libraries for discovery of inhibitors of protein–protein interactions.
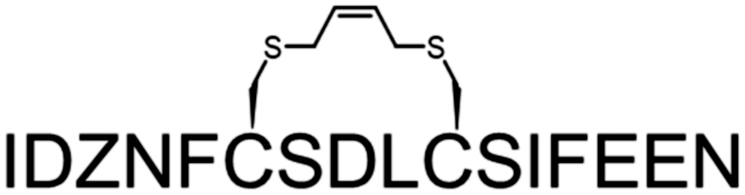
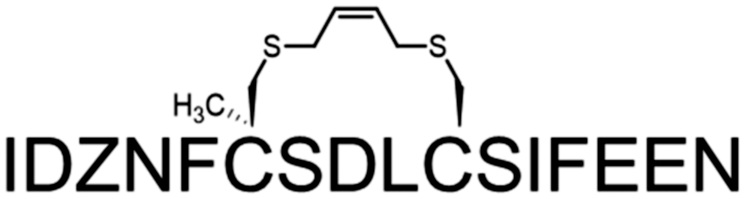
A common outcome among studies attempting to replace a hydrocarbon staple with cysteine-bis-alkylation chemistry is a loss in affinity21,25,26 and/or potency.27 We surmised that this may be due in part to loss of the α-Me group, so we set out to determine the consequences of replacing a hydrocarbon stapling residue with alpha-methyl Cys. We focused on two cysteine-bisalkylation reagents commonly applied to libraries, m-dibromoxylene (DBX)21,28–32 and cis-1,4-dichloro-2-butene (CDB).21 We chose the interaction of the human papillomavirus 16 (HPV16) E2 protein and human bromodomain 4 (BRD4) as our target. HPV is the causative agent of genital warts and a number of ano-genital and oral cancers.33 This particular protein–protein interaction is essential for HPV viral replication and transcriptional activation,34,35 and inhibitors of this interaction could potentially be therapeutics as they would disrupt the viral life cycle. The crystal structure of HPV16 E2 and a helical peptide from the C-terminal domain of BRD4 was solved by Abbate et al.36 The linear peptide derived from BRD4, 1, showed no detectable binding to HPV16-E2 protein at concentrations of up to 100 μM as evaluated by fluorescence polarization (Table 1). However, when we created a hydrocarbon stapled peptide by replacing two of the residues on the non-binding surface with a stapling residue (S)-2-(4-pentenyl)alanine, followed by cyclization, the KD was 5.6 μM (Table 1 and Fig. S1 and S2, ESI†). We then replaced these stapling residues with: two cysteines; one cysteine and one α-MeCys positioned towards the N or C-terminus; or two α-MeCys residues. The Fmoc-S-trityl-Cα-methyl cysteine-OH for the synthesis was prepared as described using a strategy based on enzymatic enrichment of a malonate half-ester.37–39 Each of these peptides was cyclized with either DBX or CDB and purified by HPLC (Table 1 and Fig. S1 and S2, ESI†).
| Name | Sequence | K D (μM ± S.D.) | |
|---|---|---|---|
| 1 | Lin |
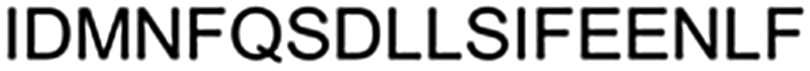
|
>100 |
| 2 | Stp |

|
5.64 ± 0.05 |
| 3 | DCB |
|
60.5 ± 0.8 |
| 4 | DCB-NMe |
|
36.9 ± 0.5 |
| 5 | DCB-CMe |

|
56.23 ± 0.13 |
| 6 | DCB-Me2 |

|
39.4 ± 0.6 |
| 7 | DBX |

|
37.1 ± 0.2 |
| 8 | DBX-NMe |

|
17.31 ± 0.12 |
| 9 | DBX-CMe |

|
29.22 ± 0.16 |
| 10 | DBX-Me2 |
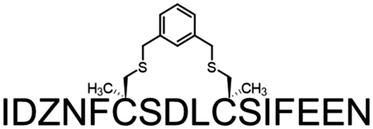
|
11.23 ± 0.08 |
These data show some interesting trends (Table 1). First, the DBX-cyclized peptides (peptides 7–10) were of higher affinity than the CDB-cyclized peptides (peptides 3–6), even though the CDB cycle is sterically most similar to the hydrocarbon staple. Second, in each case, replacing even a single Cys with α-MeCys improved the affinity. Third, placing the α-MeCys residue at the N-terminal Cys position (peptides 4, 8) was more beneficial than substitution at the C-terminal (peptides 5, 9). Finally, the doubly substituted α-MeCys peptides showed the greatest enhancement in affinity, and in the case of the DBX cycle (peptide 10), this affinity approached that of the hydrocarbon stapled peptide (peptide 2). The small magnitude of this difference in affinity is notable, as other studies with peptides comparing hydrocarbon stapling to cysteine-bis-alkylation have shown significant negative impacts to affinity.21,26 We also used circular dichroism to investigate the effects of the α-MeCys substitution and cyclization on the helicity of the non-fluorescently labeled versions of the peptides in aq. buffer with TFE (Fig. S3 and S4, ESI†). As expected, both α-MeCys incorporation and cyclization improved the helicity of the peptides, with peptide 10 showing the greatest helicity. This data highlights the importance of the α-Me substituent and DBX cyclization for creation of helicity and maintenance of affinity when moving from hydrocarbon staples to other macrocyclization strategies.
Encouraged by the promising effect of α-MeCys, we sought to develop a strategy to incorporate this AA into in vitro translated peptide libraries. This would open access to mRNA-displayed peptide libraries containing trillions of these staple-mimetic peptides for the discovery of new stapled peptide inhibitors. We recently described a promiscuous editing-deficient aminoacyl-tRNA synthetase (ValRS T222P)40 that was able to incorporate α-MeSer and α-MeAla. Since this synthetase is also able to charge cysteine onto tRNAVal,41 we posited that it would be able to charge α-MeCys. Indeed, using a MALDI AARS charging assay,42 we observed that this is the case (Fig. S5, ESI†). Using a custom version of the PURE in vitro translation system,43 lacking Val and ValRS but containing tRNAVal pre-charged with α-MeCys, we were able to achieve efficient incorporation of α-MeCys into a template containing one (Fig. S5, ESI†) or two valine codons spaced at the i, i + 4 positions (Fig. 1A, B and Fig. S6, ESI†). We note that efficient incorporation required supplementing the in vitro translation system with in vitro transcribed tRNAVal charged with α-MeCys as was the case for other α-Me AAs.40 This peptide could also be post-translationally cyclized with DBX (Fig. 1C).
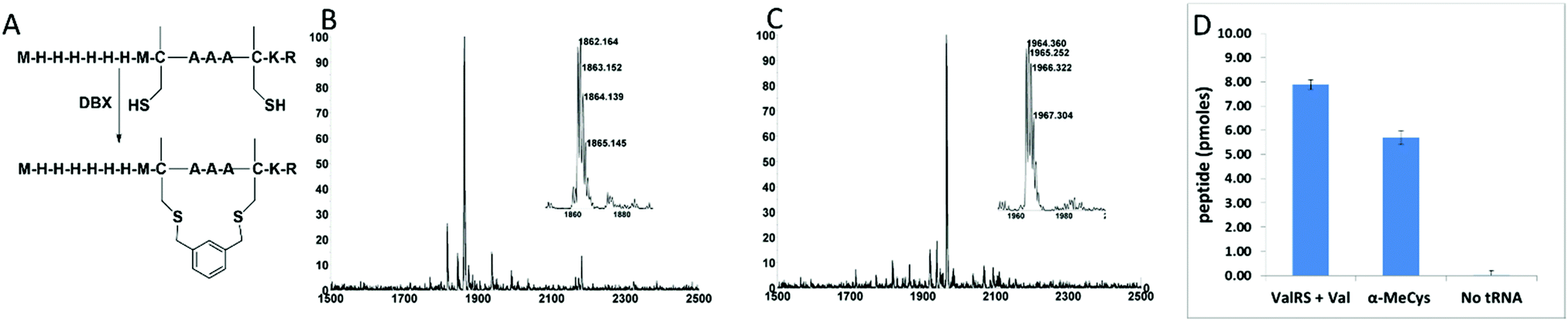 | ||
| Fig. 1 Incorporation of α-MeCys by in vitro translation. (A) Sequence and cyclization of in vitro translated peptides containing α-MeCys. (B) MALDI-TOF MS spectrum of an in vitro translation of a peptide containing two α-MeCys residues. Expected [M + H]+: 1862.80. Observed [M + H]+: 1862.16. (C) MALDI-TOF MS spectrum after cyclization of the peptide in (B) with DBX. Expected [M + H]+: 1964.85. Observed [M + H]+: 1964.36. (D) Peptide yield of an in vitro translation reaction (50 μL) of library mRNAs with two fixed valine codons in the presence of ValRS + Val, tRNAVal pre-charged with α-MeCys, or neither. The library was a mixture of three degenerate mRNAs: MXX*XXX*XXXXXXGGGH6, MXXXX*XXX*XXXXGGGH6, and MXXXXXX*XXX*XXGGGH6 where * = a fixed valine codon (GUA) and X's are randomized (NNS) positions. Yield is measured by capture onto Ni-NTA beads using 35S-Met counting after background subtraction of a control reaction lacking mRNA. Error bars represent the standard deviation from three experiments. | ||
We then tested for efficient incorporation of α-MeCys in response to a library of mRNAs (Fig. 1D). The library was designed to have fixed Val codons at i, i + 4 positions and a C-terminal His-tag. We performed three in vitro translation reactions with this library, one containing Val and ValRS, the second containing supplemented α-MeCys-tRNAVal, and the third lacking any AA-tRNA that could recognize the Val codons. The translation lacking the AA-tRNA had a very low yield, showing that readthrough of the vacated Val codons was very poor. However, both the Val/ValRS and the α-MeCys-tRNAVal translations gave high yields, demonstrating that α-MeCys can be incorporated into peptide libraries.
To demonstrate compatibility of the α-MeCys/DBX cyclization with mRNA display, we performed a round of in vitro selection combining a naïve peptide library containing fixed α-MeCys residues and a peptide derived from our highest-affinity binder (10). If the mRNA display system is working properly, this E2 binder should be captured onto E2-resin preferentially over the naïve library. Fig. 2 describes the mRNA display strategy we used to validate this.
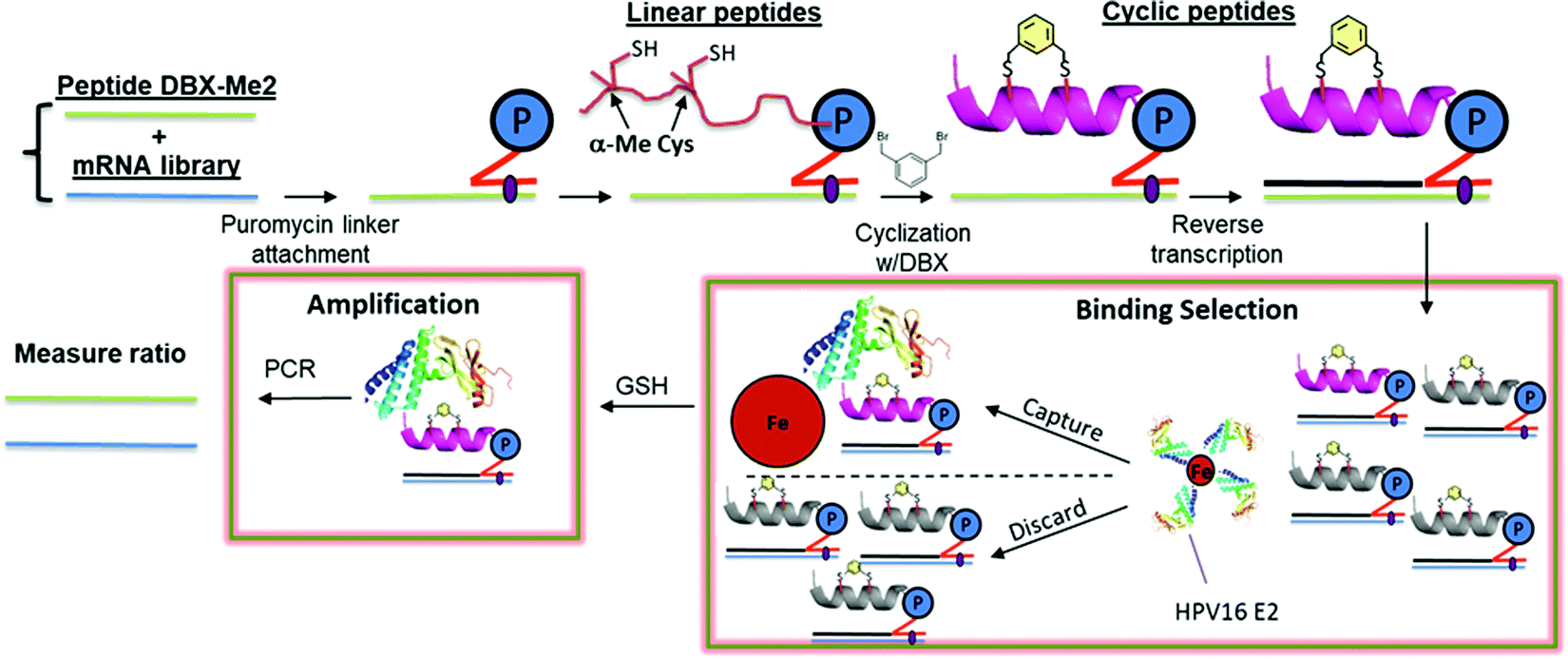 | ||
| Fig. 2 A mock selection cycle using stapled mRNA display to measure enrichment of E2 binding peptide 10. P = puromycin. mRNAs corresponding to peptide 10 and the library mRNAs were each ligated to a puromycin DNA sequence. These puromycin-mRNAs were then added to in vitro translation reactions, leading to fusion of the nascent linear peptides onto their encoding mRNAs. Each mRNA-peptide fusion was then cyclized with DBX followed by reverse transcription to generate the cDNA. The mRNA-peptide fusions were then captured onto Avi-tagged HPV16 E2.44 The non-binding mRNA-peptide fusions were washed away, and binders were then eluted with heat and amplified by PCR. | ||
To distinguish the E2-binding peptide from the naïve library, we incorporated a ClaI restriction site into the E2 binder coding sequence so that its DNA would give shorter bands on an agarose gel following ClaI digestion (Fig. 3A and B). We mixed our naïve library and the E2 binder mRNAs so that the ratio was 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Lib:E2 (1% E2) (Fig. 3B, Pre). We then took this mixture through a complete mRNA display selection cycle and measured the band intensities again (Fig. 3B, Post). After the selection, we found that the ratio had changed to 2.1:1 (32% E2), as evidenced by the darker lower bands on the gel. This demonstrates a 32-fold enrichment of our E2 binder, proving compatibility of our stapling strategy with mRNA display. A similar experiment with the non-cyclized peptide showed no enrichment (Fig. S7, ESI†).
:1 Lib:E2 (1% E2) (Fig. 3B, Pre). We then took this mixture through a complete mRNA display selection cycle and measured the band intensities again (Fig. 3B, Post). After the selection, we found that the ratio had changed to 2.1:1 (32% E2), as evidenced by the darker lower bands on the gel. This demonstrates a 32-fold enrichment of our E2 binder, proving compatibility of our stapling strategy with mRNA display. A similar experiment with the non-cyclized peptide showed no enrichment (Fig. S7, ESI†).
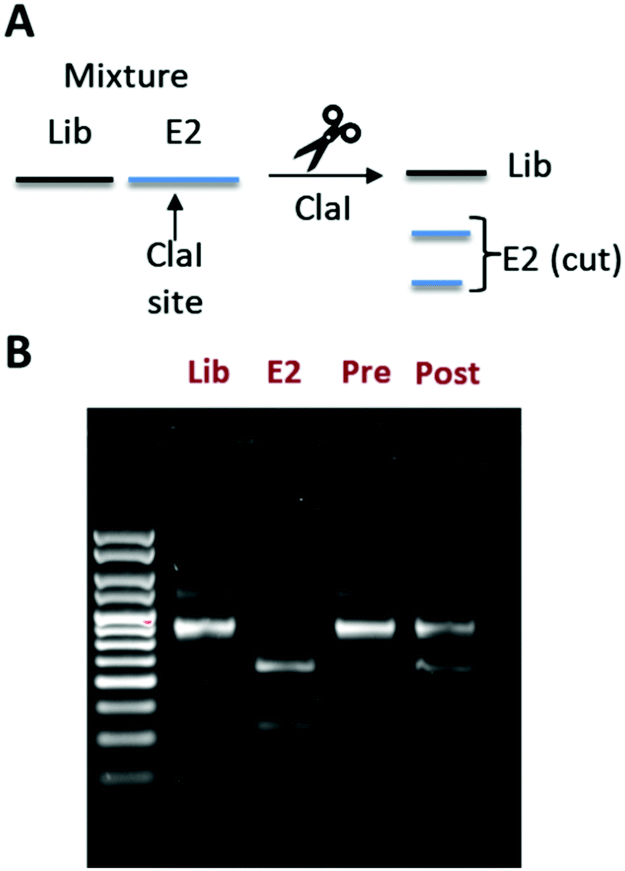 | ||
| Fig. 3 In vitro selection showing enrichment of mRNA corresponding to peptide 10. (A) Scheme illustrating how a ClaI site can be used to distinguish between DNAs encoding the naïve library (lib) or our E2-binding peptide derived from 10 (E2). (B) Agarose EtBr stained gel showing DNAs corresponding to the library (Lib) and the E2 binder (E2) alone, as well as a mixture of both before (Pre) and after (Post) one round of in vitro selection (right). The ratio of library to E2 was calculated using ImageJ by taking the top band (Lib) and dividing it by the sum of the bottom two bands (E2). | ||
Previous attempts to replace a standard hydrocarbon staple with cysteine alkylation have led to dramatic reduction in affinity. Although we also found this to be the case (Table 1), in this paper we have demonstrated that the inclusion of α-MeCys in place of cysteine can promote helicity and rescue much of this affinity loss. As such, the α-MeCys/DBX cyclization strategy promises to be interchangeable with a standard hydrocarbon staple. The compatibility of this cyclization chemistry with mRNA display opens up a new, powerful avenue for inhibitor discovery. mRNA display can create libraries that are 7 orders of magnitude larger than a typical one-bead-one-compound library and 3 orders of magnitude higher than phage-displayed libraries. Moreover, unlike phage display, mRNA display allows for the direct incorporation of ncAAs (like α-MeCys) into the peptide libraries for further optimization and enhancement of affinity. Therefore, our work initiates a new powerful strategy for discovery of inhibitors of PPIs with helical interfaces.
We are grateful to Eric Ginsburg (VCU) for his help with the circular dichroism experiments. We gratefully acknowledge the NIH (R01CA166264 to M. C. T. H., and NCI 5P30CA16059-35 for the MCC Proteomics Resource), VCU's Center for Clinical and Translational Research (to M. C. T. H. and I. M. M., NIH NCATS UL1TR002649), and the Massey Cancer Center (S. L. R.).
Conflicts of interest
M. C. T. H., E. S. I., and S. L. R. have filed for a provisional patent related to the work described herein.Notes and references
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS.
- C. Sheng, G. Dong, Z. Miao, W. Zhang and W. Wang, Chem. Soc. Rev., 2015, 44, 8238–8259 RSC.
- A. L. Jochim and P. S. Arora, ACS Chem. Biol., 2010, 5, 919–923 CrossRef CAS.
- B. N. Bullock, A. L. Jochim and P. S. Arora, J. Am. Chem. Soc., 2011, 133, 14220–14223 CrossRef CAS.
- C. Morrison, Nat. Rev. Drug Discovery, 2018, 17, 531–533 CrossRef CAS.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891–5892 CrossRef CAS.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K. H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445–E3454 CrossRef CAS.
- M. L. Stewart, E. Fire, A. E. Keating and L. D. Walensky, Nat. Chem. Biol., 2010, 6, 595–601 CrossRef CAS.
- T. N. Grossmann, J. T. Yeh, B. R. Bowman, Q. Chu, R. E. Moellering and G. L. Verdine, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17942–17947 CrossRef CAS.
- R. Rezaei Araghi, J. A. Ryan, A. Letai and A. E. Keating, ACS Chem. Biol., 2016, 11, 1238–1244 CrossRef CAS.
- D. Thean, J. S. Ebo, T. Luxton, T. Y. Yuen, F. J. Ferrer, C. W. Johannes, D. P. Lane and C. J. Brown, Sci. Rep., 2017, 7, 1763 CrossRef CAS.
- Y. V. Guillen Schlippe, M. C. T. Hartman, K. Josephson and J. W. Szostak, J. Am. Chem. Soc., 2012, 134, 10469–10477 CrossRef CAS.
- S. Li, S. Millward and R. Roberts, J. Am. Chem. Soc., 2002, 124, 9972–9973 CrossRef CAS.
- Y. Yamagishi, I. Shoji, S. Miyagawa, T. Kawakami, T. Katoh, Y. Goto and H. Suga, Chem. Biol., 2011, 18, 1562–1570 CrossRef CAS.
- D. P. Fairlie and A. Dantas de Araujo, Biopolymers, 2016, 106, 843–852 CrossRef CAS.
- H. Jo, N. Meinhardt, Y. Wu, S. Kulkarni, X. Hu, K. E. Low, P. L. Davies, W. F. DeGrado and D. C. Greenbaum, J. Am. Chem. Soc., 2012, 134, 17704–17713 CrossRef CAS.
- A. M. Spokoyny, Y. Zou, J. J. Ling, H. Yu, Y.-S. Lin and B. L. Pentelute, J. Am. Chem. Soc., 2013, 135, 5946–5949 CrossRef CAS.
- A. Muppidi, K. Doi, C. P. Ramil, H.-G. Wang and Q. Lin, Tetrahedron, 2014, 70, 7740–7745 CrossRef CAS.
- Y. Tian, Y. Jiang, J. Li, D. Wang, H. Zhao and Z. Li, ChemBioChem, 2017, 18, 2087–2093 CrossRef CAS.
- A. Muppidi, Z. Wang, X. Li, J. Chen and Q. Lin, Chem. Commun., 2011, 47, 9396 RSC.
- P. Diderich, D. Bertoldo, P. Dessen, M. M. Khan, I. Pizzitola, W. Held, J. Huelsken and C. Heinis, ACS Chem. Biol., 2016, 11, 1422–1427 CrossRef CAS.
- (a) D. J. Yeo, S. L. Warriner and A. J. Wilson, Chem. Commun., 2013, 49, 9131–9133 RSC; (b) J. A. Miles, D. J. Yeo, P. Rowell, S. Rodriguez-Marin, C. M. Pask, S. L. Warriner, T. A. Edwards and A. J. Wilson, Chem. Sci., 2016, 7, 3694–3702 RSC.
- K. Sakagami, T. Masuda, K. Kawano and S. Futaki, Mol. Pharmaceutics, 2018, 15, 1332–1340 CrossRef CAS.
- G. H. Bird, E. Mazzola, K. Opoku-Nsiah, M. A. Lammert, M. Godes, D. S. Neuberg and L. D. Walensky, Nat. Chem. Biol., 2016, 12, 845–852 CrossRef CAS.
- Y. Tian, J. Li, H. Zhao, X. Zeng, D. Wang, Q. Liu, X. Niu, X. Huang, N. Xu and Z. Li, Chem. Sci., 2016, 7, 3325–3330 RSC.
- Y. Wang and D. H. Chou, Angew. Chem., Int. Ed., 2015, 54, 10931–10934 CrossRef CAS.
- A. Muppidi, H. Zhang, F. Curreli, N. Li, A. K. Debnath and Q. Lin, Bioorg. Med. Chem. Lett., 2014, 24, 1748–1751 CrossRef CAS.
- Y. V. Guillen Schlippe, M. C. T. Hartman, K. Josephson and J. W. Szostak, J. Am. Chem. Soc., 2012, 134, 10469–10477 CrossRef CAS.
- D. E. Hacker, M. Almohaini, A. Anbazhagan, Z. Ma and M. C. T. Hartman, Methods Mol. Biol., 2015, 1248, 105–117 CrossRef CAS.
- D. E. Hacker, J. Hoinka, E. S. Iqbal, T. M. Przytycka and M. C. T. Hartman, ACS Chem. Biol., 2017, 12, 795–804 CrossRef CAS PubMed.
- E. R. White, L. Sun, Z. Ma, J. M. Beckta, B. A. Danzig, D. E. Hacker, M. Huie, D. C. Williams, R. A. Edwards, K. Valerie, J. N. M. Glover and M. C. T. Hartman, ACS Chem. Biol., 2015, 10, 1198–1208 CrossRef CAS.
- P. Timmerman, J. Beld, W. C. Puijk and R. H. Meloen, ChemBioChem, 2005, 6, 821–824 CrossRef CAS.
- H. zur Hausen, Virology, 2009, 384, 260–265 CrossRef CAS.
- M. R. Schweiger, J. You and P. M. Howley, J. Virol., 2006, 80, 4276–4285 CrossRef CAS.
- E. J. Gauson, M. M. Donaldson, E. S. Dornan, X. Wang, M. Bristol, J. M. Bodily and I. M. Morgan, J. Virol., 2015, 89, 4980–4991 CrossRef CAS.
- E. A. Abbate, C. Voitenleitner and M. R. Botchan, Mol. Cell, 2006, 24, 877–889 CrossRef CAS.
- B. L. Kedrowski, J. Org. Chem., 2003, 68, 5403–5406 CrossRef CAS.
- D. Masterson, B. Kedrowski and A. Blair, Synlett, 2010, 2941–2943 CrossRef CAS.
- H. K. Kotapati, D. R. Lawrence, S. O. Thames and D. S. Masterson, Tetrahedron Lett., 2016, 57, 4389–4391 CrossRef CAS.
- E. S. Iqbal, K. K. Dods and M. C. T. Hartman, Org. Biomol. Chem., 2018, 16, 1073–1078 RSC.
- V. Doring, H. D. Mootz, L. A. Nangle, T. L. Hendrickson, V. de Crecy-Lagard, P. Schimmel and P. Marliere, Science, 2001, 292, 501–504 CrossRef CAS.
- M. C. T. Hartman, K. Josephson and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4356–4361 CrossRef CAS.
- Y. Shimizu, A. Inoue, Y. Tomari, T. Suzuki, T. Yokogawa, K. Nishikawa and T. Ueda, Nat. Biotechnol., 2001, 19, 751–755 CrossRef CAS.
- W. Boner, E. R. Taylor, E. Tsirimonaki, K. Yamane, M. S. Campo and I. M. Morgan, J. Biol. Chem., 2002, 277, 22297–22303 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc10192b |
| This journal is © The Royal Society of Chemistry 2019 |