
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The two isomers of a cyclometallated palladium sensitizer show different photodynamic properties in cancer cells†
Xue-Quan
Zhou
 a,
Anja
Busemann
a,
Michael S.
Meijer
a,
Maxime A.
Siegler
b and
Sylvestre
Bonnet
*a
a,
Anja
Busemann
a,
Michael S.
Meijer
a,
Maxime A.
Siegler
b and
Sylvestre
Bonnet
*a
aLeiden Institute of Chemistry, Universiteit Leiden, Einsteinweg 55 2333 CC, Leiden, The Netherlands. E-mail: bonnet@chem.leidenuniv.nl
bDepartment of Chemistry, Johns Hopkins University, Maryland 21218, Baltimore, USA
First published on 18th March 2019
Abstract
This report demonstrates that changing the position of the carbon–metal bond in a polypyridyl cyclopalladated complex, i.e. going from PdL1 (N^N^C^N) to PdL2 (N^N^N^C), dramatically influences the photodynamic properties of the complex in cancer cells. This effect is primarily attributed to the significantly difference in absorbance and singlet oxygen quantum yields between the two isomers.
The success of cisplatin, a milestone drug in the treatment of cancers, stimulated the generation of many platinum-based anticancer drugs,1–3 three of which (carboplatin, oxaliplatin and nedaplatin) are approved worldwide. However, the unselective covalent binding of cisplatin with DNA in cancer cells and healthy cells results in serious side effects and drug resistances, which has encouraged the development of anticancer drugs based on alternative metals.4–9 In this regard, palladium(II) complexes have been proposed as potential metal-based anticancer drugs for their similar d8 coordination sphere and square-planar structure, compared to platinum(II) complexes.10,11 One of them, called padeliporfin or WST11, was recently approved for photodynamic therapy (PDT) of prostate cancer.12 PDT is a form of light-activated anticancer treatment. It emerges as a more patient-friendly approach due to the controlled toxicity effect and low invasiveness of light irradiation.13–17 In PDT, a photosensitizing agent (PS) is irradiated by visible light at the tumor site, where it generates cytotoxic reactive oxygen species (ROS), which induces cancer cell death.18 Polypyridyl metal complexes typically form excellent PDT sensitizers, provided they strongly absorb visible light.19,20 The light absorption properties of such complexes can be tuned by changing the metal or the ligands. Critically, good photosensitizers should be photostable, which can be achieved using multidentate ligands. The single coordination bonds in multidentate complexes are no stronger than those of monodentate ligands, they are simply less likely to all decoordinate at once.
Recently, bioactive pincer palladium complexes with tridentate N-heterocyclic carbene ligands have been shown to possess stable metal–carbon bonds and tuneable physicochemical properties.10,21–24 However, intracellular substitution of the remaining monodentate ligands makes their speciation in biological media and mode-of-action complicated to understand. In addition, due to the smaller ionic radius of Pd2+ ions, Pd–ligand bonds are longer and more labile than their Pt–ligand analogues,25 so that anticancer drugs based on palladium(II) are still comparatively rare.6 To overcome these drawbacks, we investigated the design and properties of palladium(II) PDT sensitizers built from single tetradentate cyclometallating ligands, which are expected to be more stable in biological media compared with the tridentate N-heterocyclic carbene ligands. Cyclometallation was considered for different reasons. Firstly, the strong Pd–C bond can stabilize these compounds in biological media. Secondly, the lower charge introduced by the cyclometallated ligand can improve the lipophilicity and cellular uptake of the metal complexes.7,26 Finally, the presence of a Pd–C bond should in principle lead to a bathochromic shift of the visible absorption bands of the metal complex, which is key for PDT applications.27
In polypyridyl metal complexes, introducing a metal–carbon bond usually generates a series of isomers that might have different properties. Herein we investigated two novel cyclopalladated isomers PdL1 (H2L1 = N-(3-(pyridin-2-yl)phenyl)-[2,2′-bipyridin]-6-amine) and PdL2 (H2L2 = N-(6-phenylpyridin-2-yl)-[2,2′-bipyridin]-6-amine) (Scheme 1a). In PdL1, the Pd–C bond was introduced in a pyridyl group that is adjacent to the non-bonded nitrogen bridge of the ligand, while in PdL2 it is introduced in one of the terminal aromatic rings. The ligands H2L1 and H2L2 were synthesized by Buchwald–Hartwig coupling reactions (Scheme S1 and Fig. S1–S4, ESI†).28–30 Palladation was achieved in more than 90% yield by reacting the corresponding ligand with palladium(II) acetate in acetic acid (Scheme S1 and Fig. S5–S8, ESI†). Neither 1H NMR (Fig. S5 and S7, ESI†) nor infrared spectroscopy (Fig. S9, ESI†) showed any sign of a protonated secondary amine bridge, which altogether suggested that these complexes were much more acidic than expected. According to 13C-APT NMR (Fig. S2, S4, S6 and S8, ESI†), the ligands H2L1 and H2L2 have six quaternary carbon peaks, while their palladium complexes have seven, thus demonstrating that cyclometallation did occur. Altogether PdL1 and PdL2 appear to be neutral complexes; their identical HRMS data also demonstrated they are coordination isomers.
 | ||
| Scheme 1 (a) Synthesis of PdL1 and PdL2; (b) displacement ellipsoid plot (50% probability level) of PdL2 at 110(2) K (bond distance: Pd–N1 2.060(3) Å, Pd–N2 2.028(4) Å, Pd–N4 1.988(3) Å, Pd–C21 2.017(4) Å); angle: N4–Pd1–C21 81.99(15)°, N4–Pd1–N2 92.66(16)°, C21–Pd1–N2 174.65(18)°, N4–Pd1–N1 172.2(2)°, C21–Pd1–N1 105.02(17)°, N2–Pd1–N1 80.33(13)°. | ||
Vapor diffusion of diethyl ether into a methanol solution of PdL2 yielded red rectangular crystals suitable for X-ray structure determination (Table S1, ESI,† and Scheme 1b). PdL2 crystallized in the centrosymmetric P21/n monoclinic space group. Three nitrogen and one carbon atom were coordinated to the palladium(II) cation, with bond lengths in the range 1.988(3)–2.028(4) Å for the three Pd–N bonds, and a Pd–C bond distance of 2.017(4) Å. The coordination sphere was slightly distorted, with a dihedral angle N1–N2–N4–C21 of 2.33°. τ4, a structural parameter calculated by (360° − (α + β))/(141°), where α and β are the two greatest valence angles of the coordination sphere,31 was 0.093 in the structure of PdL2, which is typical of an essentially square planar complex. Deprotonation of the nitrogen bridge was evident from the shorter distance between the amine nitrogen atoms and the adjacent pyridine carbon atoms (C10–N3 = 1.353(4) Å and C11–N3 = 1.349(4) Å), compared to that found in metal complexes with protonated nitrogen bridges (N–C distances in the range 1.36 Å to 1.39 Å).29,30 Also, unlike for [Fe(Hbbpya)(NCS)2],32 no residual electron density was found near the bridging N atom in the structure of PdL2. Finally, the asymmetric unit contained no counter-ions. In summary, single crystal X-ray crystallography is consistent with NMR and IR data, showing that PdL1 and PdL2 are neutral species because deprotonation of the nitrogen bridge becomes very easy upon coordination.
The absorption spectrum of both complexes in PBS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO (1:1) solution at 310 K (Fig. S10, ESI†) presented no significant changes over 24 hours, suggesting that the complexes were thermally stable under such conditions. Similar results were obtained in cell-growing medium (Fig. S11, ESI†), demonstrating good stability under such conditions. The partition coefficients (logPow) of the palladium complexes were determined by the shake-flask method (Table S2, ESI†). logPow was lower for PdL1 (−0.64) than for PdL2 (+0.046), confirming the higher solubility in water of the former, compared to the latter. Their cytotoxicity was tested in lung (A549) and skin (A431) cancer cell lines, both in the dark and upon blue light activation. Low doses of blue light were chosen (455 nm, 5 min, 10.5 mW cm−2, 3.2 J cm−2) which have by themselves no effect on cell growth.33 The cell growth inhibition effective concentrations (EC50) of PdL1 and PdL2 are reported in Table 1, and the dose–response curves are shown in Fig. 1 and Fig. S12 (ESI†). In the dark both compounds showed significant anticancer activity, with an EC50 around 10 μM for PdL1 and PdL2 in A549 cells, respectively. After blue light activation, PdL1 showed a notable 13- or 4.0-fold increase in cytotoxicity in A549 and A431, respectively, while PdL2 showed a negligible photoindex of 1.3 or 1.4, respectively. The difference in photocytotoxicity between the two coordination isomers was quite intriguing. To investigate the reason for such a difference, we first measured the singlet oxygen (1O2) generation quantum yield (φΔ) of these two isomers in CD3OD spectroscopically. φΔ was more than twice higher for PdL1 (0.89) than for PdL2 (0.38, Fig. 2b and Table S3, ESI†), and higher than the reference [Ru(bpy)3]Cl2 (0.73).34 However, PdL2 was still a decent 1O2 generator. In methanol, the absorbance spectra of both complexes (Fig. 2) were similar in the 270–300 nm region; however, PdL1 had a much higher absorption in the blue region with λabsmax = 422 nm, compared to PdL2 that absorbed in the near-UV region (λabsmax = 347 nm). In this solvent the molar absorptivity at 455 nm for PdL1 and PdL2 was 2004 M−1 cm−1 and 133 M−1 cm−1, respectively, indicating a 15-fold enhanced absorption of PdL1 in the blue region, compared with PdL2. Considering their similar lifetime (0.271 vs. 0.333 ns for the main component of their biexponential decay, Table S3 and Fig. S13, ESI†), the difference in 1O2 generation efficiency is probably a consequence of the higher phosphorescence quantum yield for PdL1 (0.0017) vs.PdL2 (0.00084, Table S3, ESI†), which points to the slower non-radiative decay pathways for the former, compared to the latter. Altogether, the dramatically higher phototoxicity of PdL1, compared to PdL2, seems to result from the much better absorption of blue light of PdL1, coupled to its higher phosphorescence quantum yield, which leads to higher 1O2 generation efficiency. Although different logPow values may lead to different cell uptake and sub-cellular localization for both isomers as well, the better photobiological properties of PdL1 depend, at least in part, on the much better photodynamic properties of PdL1 (ε455 and φΔ), compared to its isomer PdL2.
:DMSO (1:1) solution at 310 K (Fig. S10, ESI†) presented no significant changes over 24 hours, suggesting that the complexes were thermally stable under such conditions. Similar results were obtained in cell-growing medium (Fig. S11, ESI†), demonstrating good stability under such conditions. The partition coefficients (logPow) of the palladium complexes were determined by the shake-flask method (Table S2, ESI†). logPow was lower for PdL1 (−0.64) than for PdL2 (+0.046), confirming the higher solubility in water of the former, compared to the latter. Their cytotoxicity was tested in lung (A549) and skin (A431) cancer cell lines, both in the dark and upon blue light activation. Low doses of blue light were chosen (455 nm, 5 min, 10.5 mW cm−2, 3.2 J cm−2) which have by themselves no effect on cell growth.33 The cell growth inhibition effective concentrations (EC50) of PdL1 and PdL2 are reported in Table 1, and the dose–response curves are shown in Fig. 1 and Fig. S12 (ESI†). In the dark both compounds showed significant anticancer activity, with an EC50 around 10 μM for PdL1 and PdL2 in A549 cells, respectively. After blue light activation, PdL1 showed a notable 13- or 4.0-fold increase in cytotoxicity in A549 and A431, respectively, while PdL2 showed a negligible photoindex of 1.3 or 1.4, respectively. The difference in photocytotoxicity between the two coordination isomers was quite intriguing. To investigate the reason for such a difference, we first measured the singlet oxygen (1O2) generation quantum yield (φΔ) of these two isomers in CD3OD spectroscopically. φΔ was more than twice higher for PdL1 (0.89) than for PdL2 (0.38, Fig. 2b and Table S3, ESI†), and higher than the reference [Ru(bpy)3]Cl2 (0.73).34 However, PdL2 was still a decent 1O2 generator. In methanol, the absorbance spectra of both complexes (Fig. 2) were similar in the 270–300 nm region; however, PdL1 had a much higher absorption in the blue region with λabsmax = 422 nm, compared to PdL2 that absorbed in the near-UV region (λabsmax = 347 nm). In this solvent the molar absorptivity at 455 nm for PdL1 and PdL2 was 2004 M−1 cm−1 and 133 M−1 cm−1, respectively, indicating a 15-fold enhanced absorption of PdL1 in the blue region, compared with PdL2. Considering their similar lifetime (0.271 vs. 0.333 ns for the main component of their biexponential decay, Table S3 and Fig. S13, ESI†), the difference in 1O2 generation efficiency is probably a consequence of the higher phosphorescence quantum yield for PdL1 (0.0017) vs.PdL2 (0.00084, Table S3, ESI†), which points to the slower non-radiative decay pathways for the former, compared to the latter. Altogether, the dramatically higher phototoxicity of PdL1, compared to PdL2, seems to result from the much better absorption of blue light of PdL1, coupled to its higher phosphorescence quantum yield, which leads to higher 1O2 generation efficiency. Although different logPow values may lead to different cell uptake and sub-cellular localization for both isomers as well, the better photobiological properties of PdL1 depend, at least in part, on the much better photodynamic properties of PdL1 (ε455 and φΔ), compared to its isomer PdL2.
| Complexes | EC50 (μM) | ||||
|---|---|---|---|---|---|
| A549 | ±CI | A431 | ±CI | ||
| Irradiation condition: 455 nm blue light, 5 min, 10.5 mW cm−2, 3.2 J cm−2. Data is the mean over three independent experiments. | |||||
| PdL1 | Dark | 12 | +3.0 | 20 | +4.0 |
| −3.0 | −3.0 | ||||
| Light | 0.9 | +0.8 | 5.0 | +2.0 | |
| −0.5 | −1.0 | ||||
| PI | 13 | 4.0 | |||
| PdL2 | Dark | 8.0 | +2.0 | 14 | +2.0 |
| −1.0 | −1.0 | ||||
| Light | 6.0 | +0.8 | 10 | +1.0 | |
| −0.7 | −1.0 | ||||
| PI | 1.3 | 1.4 | |||
 | ||
| Fig. 1 Dose–response curves for A549 cells incubated with palladium complexes and irradiated 5 min with blue light (blue data points), or in the dark (black data points). | ||
 | ||
| Fig. 2 (a) The molar absorption coefficient (solid line) and emission spectra (dashed line) of PdL1 (black), PdL2 (red) in CH3OH. (b) Singlet oxygen emission spectra of [Ru(bpy)3]Cl2 (blue), PdL1 (black), PdL2 (red) in CD3OD irradiated with blue light (λex = 450 nm, 50 mW, 0.4 W cm−2). | ||
Density functional theory (DFT) calculations were performed to understand why PdL1 exhibited higher absorption in the blue domain than its isomer PdL2. The nature of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbitals (LUMO) is highly relevant to predict the photophysical properties of metal complexes.34–36 As showed in Fig. 3, the HOMO and LUMO orbitals of both isomers PdL1 and PdL2 had π symmetry and were centered on the ligand, with a negligible contribution of the palladium(II) center. The bridged secondary amine is the major contributor to the HOMO of both palladium complexes, attributing for 20.4 (PdL1) and 21.8% (PdL2) of the electron density. The rest of the HOMO orbital density was located in the aromatic rings directly connected to the nitrogen bridge. By contrast, the LUMO orbitals for both complexes were centered on the bipyridyl fragment. This suggested that the lowest energy absorption band of both palladium complexes should be of ligand-to-ligand charge transfer character, from the amine to the bipyridyl.
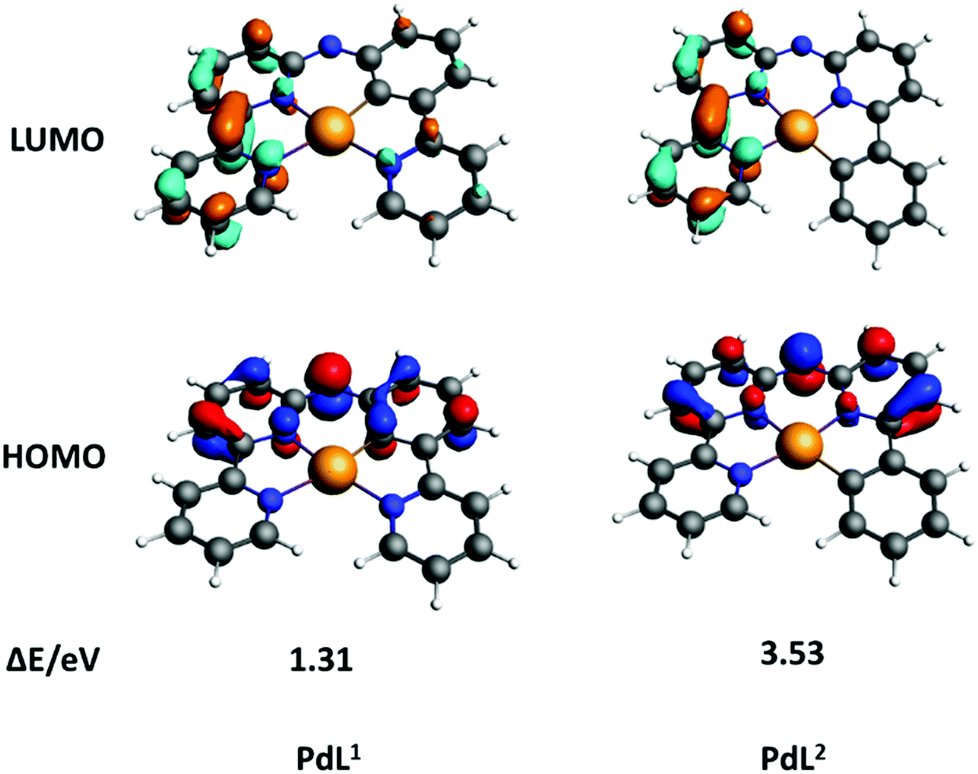 | ||
| Fig. 3 DFT calculation of HOMOs (bottom) and LUMOs (top) orbitals of PdL1 and PdL2; occupied orbitals (HOMO) have red and blue lobes, and unoccupied orbitals (LUMO) brown and cyan lobes. Element colour code: grey = C; orange = Pd; blue = N; white = H. | ||
The calculated energies of HOMOs, LUMOs and energy gaps are listed in Table S4 (ESI†). The HOMO of PdL1 was significantly higher in energy than that of PdL2, indicating the higher electron-donating effect of the negatively charged carbon atom of PdL1, compared with that of PdL2 which is further away from the nitrogen bridge. By contrast, the LUMO energy levels of both palladium complexes were similar, because LUMO orbitals are located on the almost equivalent bipyridyl fragments. Such lower energy gap of PdL1 suggested better absorption of low-energy light, which explains the observed differences in the UV-vis spectra of the two isomers. These results were confirmed by time-dependent density functional theory calculations (TDDFT) for both complexes in methanol, using COSMO to simulate solvent effects (Fig. S14, ESI,† left). The calculated spectrum of PdL1 (Fig. S14, ESI,† left) showed a lower energy (515 nm) for the HOMO–LUMO transition, compared to PdL2 (449 nm). These transition energies were increased (404 and 367 nm, respectively) by protonation of the nitrogen bridge (Fig. S14, ESI,† right), which may happen in the slightly acidic environment of cancer cells; however, the trend between [Pd(HL1)]+ and [Pd(HL2)]+ was identical to that seen for PdL1 and PdL2. Overall, calculations clearly demonstrated that a change of the position of the carbon–metal bond had a strong influence on the HOMO–LUMO energy gaps of these cyclometallated palladium complexes.
In summary, the new cyclopalladated complex PdL1 showed good absorbance in the blue region of the spectrum, low phosphorescence, and excellent singlet oxygen quantum yield (0.89), which altogether translated into high photoindex in human cancer cells. By contrast, its isomer PdL2 had low absorption and low singlet oxygen quantum yield (0.38), resulting in negligible activation by blue light in vitro. DFT calculation showed that the higher absorption in the blue region of PdL1, and thus its lower HOMO–LOMO energy gap, was due to the closer proximity between the electron-rich cyclometallated aromatic cycle and the nitrogen bridge of the ligand, while in PdL2 both aromatic rings adjacent to the N bridge are electron-poor pyridine rings, which lowers the HOMO energy level. To the best of our knowledge, this study is the first report that two isomers of organometallic prodrugs have different photobiological properties. These results demonstrate that changing the position of the carbon–metal bond in the coordination sphere of photoactive organometallic prodrugs can be used to tune the energy gap between their frontier orbitals, and hence their absorption in the visible region of the spectrum.
X. Zhou gratefully acknowledges the China Scholarship Council (CSC) for a personal grant (No. 201606200045). This work is supported by an ERC Starting Grant to S. Bonnet.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- L. Ma, N. Wang, R. Ma, C. Li, Z. Xu, M. K. Tse and G. Zhu, Angew. Chem., Int. Ed., 2018, 57, 1–6 CrossRef.
- S. Medici, M. Peana, V. M. Nurchi, J. I. Lachowicz, G. Crisponi and M. A. Zoroddu, Coord. Chem. Rev., 2015, 284, 329–350 CrossRef CAS.
- N. Cutillas, G. S. Yellol, C. de Haro, C. Vicente, V. Rodriguez and J. Ruiz, Coord. Chem. Rev., 2013, 257, 2784–2797 CrossRef CAS.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z. S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- H. Huang, P. Zhang, H. Chen, L. Ji and H. Chao, Chem. – Eur. J., 2015, 21, 715–725 CrossRef CAS PubMed.
- T. T. Fong, C. N. Lok, C. Y. Chung, Y. M. Fung, P. K. Chow, P. K. Wan and C. M. Che, Angew. Chem., Int. Ed., 2016, 55, 11935–11939 CrossRef CAS PubMed.
- M. Fanelli, M. Formica, V. Fusi, L. Giorgi, M. Micheloni and P. Paoli, Coord. Chem. Rev., 2016, 310, 41–79 CrossRef CAS.
- A.-R. Azzouzi, S. Vincendeau, E. Barret, A. Cicco, F. Kleinclauss, H. G. van der Poel, C. G. Stief, J. Rassweiler, G. Salomon, E. Solsona, A. Alcaraz, T. T. Tammela, D. J. Rosario, F. Gomez-Veiga, G. Ahlgren, F. Benzaghou, B. Gaillac, B. Amzal, F. M. J. Debruyne, G. Fromont, C. Gratzke and M. Emberton, Lancet Oncol., 2017, 18, 181–191 CrossRef CAS PubMed.
- H. Cao, L. Wang, Y. Yang, J. Li, Y. Qi, Y. Li, Y. Li, H. Wang and J. Li, Angew. Chem., Int. Ed., 2018, 57, 7759–7763 CrossRef CAS PubMed.
- Y. Ma, X. Li, A. Li, P. Yang, C. Zhang and B. Tang, Angew. Chem., Int. Ed., 2017, 56, 13752–13756 CrossRef CAS PubMed.
- S. H. Askes, A. Bahreman and S. Bonnet, Angew. Chem., Int. Ed., 2014, 53, 1029–1033 CrossRef CAS PubMed.
- S. L. Higgins and K. J. Brewer, Angew. Chem., Int. Ed., 2012, 51, 11420–11422 CrossRef CAS PubMed.
- H. Bi, Y. Dai, P. Yang, J. Xu, D. Yang, S. Gai, F. He, B. Liu, C. Zhong, G. An and J. Lin, Small, 2018, 14, e1703809 CrossRef PubMed.
- H. Bi, Y. Dai, P. Yang, J. Xu, D. Yang, S. Gai, F. He, G. An, C. Zhong and J. Lin, Chem. Eng. J., 2019, 356, 543–553 CrossRef CAS.
- J. D. Knoll and C. Turro, Coord. Chem. Rev., 2015, 282-283, 110–126 CrossRef CAS PubMed.
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed.
- J.-Y. Lee, J.-Y. Lee, Y.-Y. Chang, C.-H. Hu, N. M. Wang and H. M. Lee, Organometallics, 2015, 34, 4359–4368 CrossRef CAS.
- S. Ray, R. Mohan, J. K. Singh, M. K. Samantaray, M. M. Shaikh, D. Panda and P. Ghosh, J. Am. Chem. Soc., 2007, 15042–15053 CrossRef CAS PubMed.
- S. G. Churusova, D. V. Aleksanyan, E. Y. Rybalkina, O. Y. Susova, V. V. Brunova, R. R. Aysin, Y. V. Nelyubina, A. S. Peregudov, E. I. Gutsul, Z. S. Klemenkova and V. A. Kozlov, Inorg. Chem., 2017, 56, 9834–9850 CrossRef CAS PubMed.
- W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC.
- J. Ruiz, V. Rodriguez, C. de Haro, A. Espinosa, J. Perez and C. Janiak, Dalton Trans., 2010, 39, 3290–3301 RSC.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- S. Bonnet, Comments Inorg. Chem., 2014, 35, 179–213 CrossRef.
- Z. Fan, J. Ni and A. Zhang, J. Am. Chem. Soc., 2016, 138, 8470–8475 CrossRef CAS PubMed.
- V. H. S. van Rixel, B. Siewert, S. L. Hopkins, S. H. C. Askes, A. Busemann, M. A. Siegler and S. Bonnet, Chem. Sci., 2016, 7, 4922–4929 RSC.
- E. M. Hernández, S. Zheng, H. J. Shepherd, D. S. Yufit, K. Ridier, S. Bedoui, W. Nicolazzi, V. Velázquez, S. Bonnet, G. Molnár and A. Bousseksou, J. Phys. Chem. C, 2016, 120, 27608–27617 CrossRef.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- S. Zheng, N. R. Reintjens, M. A. Siegler, O. Roubeau, E. Bouwman, A. Rudavskyi, R. W. Havenith and S. Bonnet, Chem. – Eur. J., 2016, 22, 331–339 CrossRef CAS PubMed.
- S. L. Hopkins, B. Siewert, S. H. Askes, P. Veldhuizen, R. Zwier, M. Heger and S. Bonnet, Photochem. Photobiol. Sci., 2016, 15, 644–653 RSC.
- X. Li, J. Zhang, Z. Zhao, L. Wang, H. Yang, Q. Chang, N. Jiang, Z. Liu, Z. Bian, W. Liu, Z. Lu and C. Huang, Adv. Mater., 2018, 30, e1705005 CrossRef PubMed.
- F. F. Hung, S. X. Wu, W. P. To, W. L. Kwong, X. Guan, W. Lu, K. H. Low and C. M. Che, Chem. – Asian J., 2017, 12, 145–158 CrossRef CAS PubMed.
- J. Fernandez-Cestau, B. t. Bertrand, A. Pintus and M. Bochmann, Organometallics, 2017, 36, 3304–3312 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, graphical results, and videos. CCDC 1534260. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc10134e |
| This journal is © The Royal Society of Chemistry 2019 |