
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing membrane asymmetry of ABC polymersomes†
Evgeniia V.
Konishcheva
*ab,
Davy
Daubian
a,
Serena
Rigo
a and
Wolfgang P.
Meier
 *a
*a
aDepartment of Physical Chemistry, University of Basel, Mattenstrasse 24a, BPR 1096, 4058 Basel, Switzerland. E-mail: wolfgang.meier@unibas.ch
bPrecision Macromolecular Chemistry, Institute Charles Sadron, UPR-22 CNRS, BP 84047, 23 rue du Loess, 67034 Strasbourg Cedex 2, France. E-mail: ev.konishcheva@gmail.com
First published on 4th January 2019
Abstract
We report the sensitivity of the membrane asymmetry of ABC (PEO-b-PCL-b-PMOXA) polymersomes towards the end-group modification of a shorter C block. While a non-modified ABC polymer formed polymersomes with the A block outside and the C block inside, a mixture of ABC and ABC-biotin formed polymersomes with the C block outside.
Polymersomes have received considerable attention due to their diverse applications, including drug delivery and artificial nanoreactors.1–5 Typically, polymersomes have a symmetric membrane because they are formed by AB6–10 or ABA12 block copolymers, where A is a soluble block and B is a non-soluble block. Polymersomes assembled from ABC block copolymers, where the A and C blocks are soluble, have an asymmetric membrane with a longer soluble block forming the outer surface, and a shorter one forming the inner surface of the polymersomes.3 Such membrane asymmetry offers several advantages for the design of sophisticated structures. First of all, membrane asymmetry is an important step towards mimicking natural asymmetric cell membranes,13,14 and ABC membranes have been shown to be beneficial for the directed insertion of transmembrane proteins.15,16 Depending on the nature of the A and C blocks, asymmetry can result in different properties of the inner and outer surface of polymersomes. For example, one of the two hydrophilic blocks can be charged, thus resulting in an asymmetric membrane carrying a charge only on one side of the membrane, which consequently leads to different affinity to proteins and enhancement of drug delivery.17–21 In addition, an asymmetric membrane can carry different functional groups on the inner and outer surfaces11 for subsequent selective modifications of either side of the membrane.
The asymmetry of ABC membranes is a consequence of the packing parameter22 (i.e., geometric shape occupied by polymer chains) and incompatibility of the two soluble blocks.11,15,23,24 It is believed that the packing parameter plays a predominant role in the formation of membrane asymmetry, and even blocks of the same nature but different lengths segregate on different sides of the membrane.25 The packing parameter is very sensitive to slight changes in the block ratio caused by the variation of hydrophilicity/hydrophobicity under external stimuli or chemical modification. For example, structures assembled from stimulus-responsive polymers may undergo order–order transitions (e.g., polymersome-to-worm, polymersome-to-micelle) upon the change of temperature26–33 or pH,34–36 in the presence of enzymes,37 by host–guest recognition38 or in the presence of a cross-linking agent,39etc. Moreover, even the modification of only end-groups already induces morphological transitions.40–42 Thus, one might expect that the packing parameter of ABC molecules, and therefore the molecule orientation within the polymersome membrane, may be affected by end-group modification. To test this hypothesis, in the present study we probe the sensitivity of the membrane asymmetry of ABC polymersomes towards the end-group modification of a shorter hydrophilic block located inside.
As a model system, we chose polymersomes formed by poly(ethylene oxide)-block-polycaprolactone-block-poly(2-methyl-2-oxazoline) (PEO45-b-PCL110-b-PMOXA4 = ABC) in an aqueous solution.11 These polymersomes have an asymmetric membrane with a longer A (PEO) block located outside and a shorter C block (PMOXA) located inside, which was proven by two independent methods. These ABC polymersomes stayed intact during at least 6 months of storage at room temperature. To increase the length of the C block by its end-group modification, a biotin moiety was attached to the ABC-N3 (PEO45-b-PCL103-b-PMOXA4-N3) polymer resulting in ABC-biotin. As a control, we synthesized biotin-ABC (biotin-PEO45-b-PCL100-b-PMOXA4). ABC polymers were synthesized via coordination-insertion ring-opening polymerization of ε-caprolactone on a PEO macroinitiator followed by ω-tosylation and subsequent cationic ring-opening polymerization of 2-methyl-2-oxazoline as described previously (see ESI†).11,43 The ABC-biotin polymer was synthesized via coupling between DBCO-biotin and ABC-N3; the biotin-ABC polymer was synthesized via Cu(I)-catalyzed azide–alkyne cycloaddition (CuAAC) between alkyne-ABC and biotin-N3 (Fig. 1).
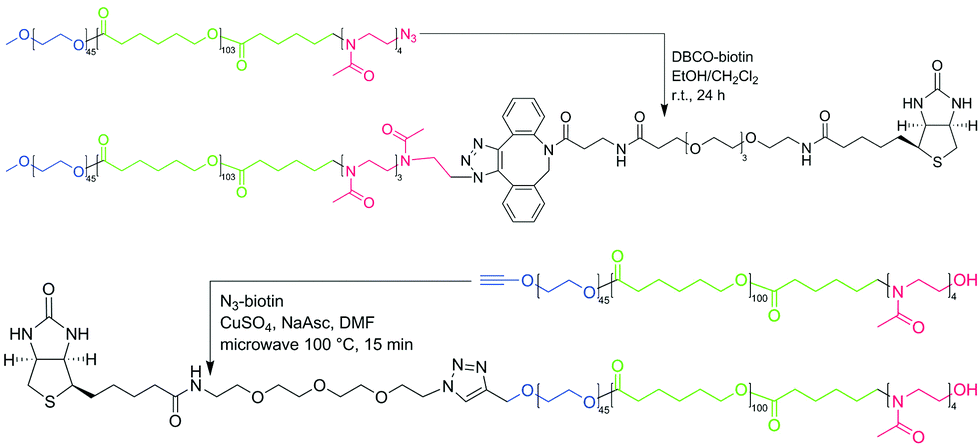 | ||
| Fig. 1 Scheme of synthesis of ABC-biotin (obtained via Cu-free DBCO – azide coupling) and biotin-ABC (obtained via microwave-assisted CuAAC) used in this study. | ||
ABC-biotin synthesis proceeded smoothly at room temperature, which was confirmed by the appearance of the peaks characteristic for biotin and disappearance of the signal from the methylene group next to azide in the 1H NMR spectrum (Fig. S1, ESI†). Attachment of biotin could not be determined quantitatively from the 1H NMR spectrum due to only partial solubility of biotin in CDCl3 or DMF-d7, which meant that the signals from this group were rather low. However, we assumed that 100% of ABC-N3 converted into ABC-biotin, because we used 5-fold excess of DBCO-biotin, and the signal from the methylene group next to the azide disappeared completely. Commercially available kits for biotin quantification were not applicable to our system, since the detection implies enzymatic reaction performed in aqueous solution.
Modification of ABC from the C (PMOXA) terminus is a relatively simple process, because the polymerization of PMOXA is the last step in the synthesis. Several functional groups can be obtained by quenching the polymerization with a specific reagent.44 Introduction of the functional group on the A (PEO) terminus is more demanding due to possible interference of such group with subsequent synthetic steps and a limited number of commercially available heterobifunctional PEO macroinitiators. We chose an alkyne-PEO-OH macroinitiator, since the alkyne group was inert in the subsequent synthetic steps, and an OH group was required for polymerization of ε-caprolactone. N3-biotin was then attached to alkyne-ABC via CuAAC, frequently used in organic chemistry and biochemistry due to high yields and a wide range of applicable conditions.45,46 However, the reaction between N3-biotin and alkyne-ABC did not proceed under different tested conditions (e.g., catalytic systems containing different Cu(I) and Cu(II) salts and ligands; T = 20–100 °C; solvents: DMF, CH2Cl2/EtOH, THF, THF/H2O; t = 1–13 days). Finally, we tested microwave-assisted synthesis, since it was shown to be advantageous for coupling of alkyne-PCL and heptakis-azido-β-cyclodextrin by Hoogenboom et al.47 Already after 15 min under microwave irradiation at 100 °C we obtained a biotin-ABC polymer. Similar to ABC-biotin, the percentage of functionalization of biotin-ABC could not be determined from integration of the signals from the biotin group on the 1H NMR spectrum, but the conversion was assumed to be 100% due to the disappearance of the signal from the methylene group next to the alkyne (data not shown).
The synthesized ABC-biotin/biotin-ABC polymers were blended with ABC at different ratios (1, 5, and 10 w/w%) prior to aqueous self-assembly using the film rehydration method at 62 °C. The final polymer concentration after self-assembly was 2 mg mL−1. The presence of biotinylated polymers did not affect the formation of polymersomes (Fig. S2, ESI†).
To detect biotin on the outer surface of polymersomes, aqueous solutions of polymersomes and Cy5-labeled streptavidin (Cy5-SA) were mixed at 20 °C prior to fluorescence correlation spectroscopy (FCS)48 experiments and laser scanning microscopy (LSM) imaging. The final Cy5-SA concentration was 0.05 mg mL−1, and the final polymer concentration was 1 mg mL−1. This corresponds to ∼1000-fold excess of biotin over Cy5-SA in the case of 1% of ABC-biotin. Such excess was chosen to ensure the efficient binding of Cy5-SA to avoid background noise in the LSM images caused by Cy5-SA in solution. In addition, the latter ratio was optimal, because the higher Cy5-SA concentration led to its aggregation, and the lower concentration of polymersomes resulted in an insufficient number of events detected during FCS measurements.
We measured aqueous solutions containing only Cy5-SA (reference), ABC polymersomes and Cy5-SA (negative control), ABC + biotin-ABC polymersomes and Cy5-SA (positive control), and ABC + ABC-biotin polymersomes and Cy5-SA. Samples containing only Cy5-SA or ABC polymersomes with Cy5-SA showed similar responses (Fig. 2a, black and green curves). The latter indicates no unspecific binding of Cy5-SA to polymersomes or its penetration inside them. The absence of unspecific binding was also confirmed by the lack of fluorescent polymersomes in LSM images (Fig. 2b). This non-interactive behavior can be attributed to the protein-repellent nature of both hydrophilic blocks (i.e., PEO and PMOXA).49,50
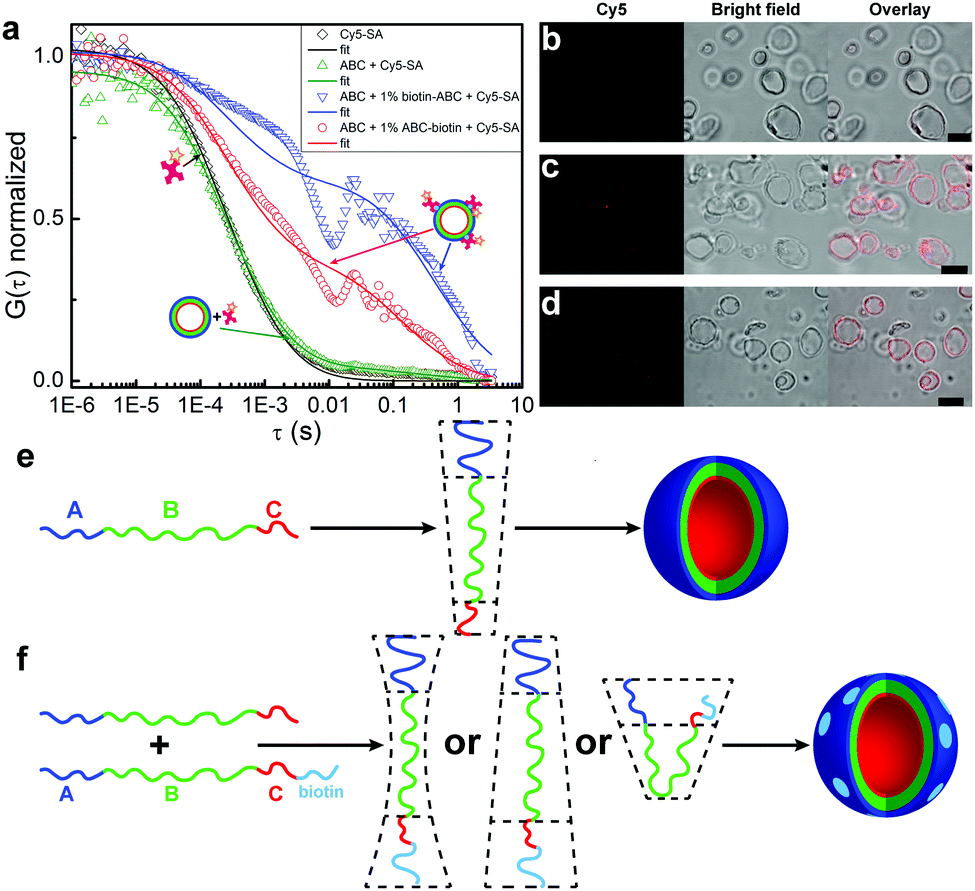 | ||
| Fig. 2 Binding between polymersomes and Cy5-SA. (a) Normalized autocorrelation curves from FCS data in water: 0.05 mg mL−1 Cy5-SA (black), 0.05 mg mL−1 Cy5-SA and 1 mg mL−1 ABC polymersomes (green curve, image b), 0.05 mg mL−1 Cy5-SA and 1 mg mL−1 polymersomes formed by ABC containing 1% of biotin-ABC (blue curve, image c), 0.05 mg mL−1 Cy5-SA and 1 mg mL−1 polymersomes formed by ABC containing 1% of ABC-biotin (red curve, image d). The LSM, bright field, and overlay images (b–d) were obtained under identical microscope settings. Scale bars are 5 μm. Schematic representation of the packing geometry of (e) ABC11 and (f) ABC-biotin molecules. Note, the actual distribution (homogenous or domain-forming) of biotin along the membrane remains unclear; the light blue domains are drawn for simplicity reason. | ||
Samples containing polymersomes with biotinylated polymers and Cy5-SA exhibited increased diffusion times (Fig. 2a, red and blue curves), which confirmed binding of the protein molecules to such polymersomes. Similar diffusion times were obtained for ABC polymersomes stained with hydrophobic Bodipy 630/650 dye (Fig. S3, ESI†). The percentage of bound Cy5-SA was ∼40–60% (Table 1) and did not depend on the amount (1%, 5%, 10%) of ABC-biotin/biotin-ABC polymers. Longer incubation time (up to 48 h) yielded similar results.
| Sample | D of polymersomes, μm | Fraction of Cy5-SA bound to polymersomes, % |
|---|---|---|
| ABC | 4.2 ± 1.6 | 4 ± 1 |
| ABC + 1% biotin-ABC | 3.4 ± 1.2 | 53 ± 18 |
| ABC + 1% ABC-biotin | 3.8 ± 1.5 | 42 ± 12 |
The presented FCS data qualitatively indicated the presence of biotin moieties on the outer surface of polymersomes, but could not be used for quantitative analysis because of the large sizes of polymersomes (Table 1). The typical confocal volume in FCS is ∼1 fL,51,52 whereas the average volume of polymersomes is ∼30 fL (see ESI†). Besides, FCS measurements were complicated by fast sedimentation and aggregation of polymersomes. As can be seen from Fig. 2a, the autocorrelation curves of the samples with ABC-biotin/biotin-ABC contain some spikes at the diffusion time >0.01 s.
The FCS data were supported by LSM imaging (Fig. 2b–d). Non-biotinylated polymersomes stayed non-fluorescent in the presence of Cy5-SA, whereas the membranes of biotinylated polymersomes became fluorescent due to the binding of Cy5-SA. The latter indicates that binding happens only between the membrane and the protein and excludes the penetration of Cy5-SA inside the polymersomes.
As can be seen from the FCS and LSM data, the attachment of biotin to the shorter C (PMOXA) block results in its appearance on the outer surface of polymersomes. The “flip” of the C block from the inside to the outside can be a consequence of geometric and/or physico-chemical factors. The geometric factor implies the change of the packing shape occupied by ABC-biotin molecules compared to ABC. The physico-chemical factor implies the favored interactions between the A and C-biotin hydrophilic blocks. For example, it could be the interaction between the A (PEO) block and PEO spacer between C and biotin in ABC-biotin polymers (Fig. 1). Regardless of which factor dominates the “flip” of the C block, the only molecules that undergo such transition should be those containing biotin, i.e., ABC-biotin.
We believe that the geometric factor plays a predominant role,25 because the counter length of the biotinylated PMOXA block (∼30 Å, 1090 g mol−1) was twice as long as that of non-biotinylated PMOXA (∼15 Å, 340 g mol−1). Our hypothesis is supported by self-assembly of pure ABC-biotin polymer: while non-modified ABC self-assembles into polymersomes (packing shape is a cylinder, Fig. 2e), pure ABC-biotin forms a mixture of polymersomes and cloud-like aggregates (double-cone packing shape, Fig. 2f and Fig. S4, ESI†).53 The packing geometry of ABC-biotin chains in I-shaped conformation is most likely intermediate between cylinders and double-cone shape. U-shaped conformation is also possible, but less likely because ABC polymersomes contain predominantly molecules in the I-shaped conformation,11 and the structures formed by ABC molecules in the U-shaped conformation (e.g., worms) were not thermodynamically stable and transformed into polymersomes.53 The conformation of ABC-biotin molecules might be investigated by measuring spatial interactions between A (PEO) and C (PMOXA) blocks by 2D nuclear Overhauser effect spectroscopy (NOESY NMR) or Förster resonance energy transfer (FRET). NOESY NMR was not applicable to our system presumably due to the large size of the polymersomes, as we have already tested earlier.11 FRET experiments imply the presence of two fluorescent dyes, donor and acceptor, on the A and C ends in one ABC-biotin molecule. In this case the final polymer should have a sequence donor-ABC-biotin-acceptor. This would not only be a very demanding synthetic procedure, but more importantly, such molecules might have a completely different orientation in the membrane, as already biotin affects the membrane asymmetry. The amount, conformation (i.e., I- or U-shape), and distribution (i.e., homogenous or domain-forming) of ABC-biotin molecules in the membrane could not be investigated with the conventional LSM or FCS due to their limitations, and thus these issues will be studied further elsewhere.
The experimental data presented here suggest that one should carefully consider the end-group modification of ABC polymers with respect to the membrane asymmetry. To conclude that the presented findings can be regarded as a general rule, other ABC systems of different chemical nature, block lengths, and polymersome size should be systematically investigated. Also, another question which should be addressed next is whether the asymmetry is influenced by in situ end-group modification, i.e., when the end-group modification is performed on already assembled polymersomes.
We acknowledge SNSF, NCCR Molecular Systems Engineering, and the University of Basel for financial support.
Conflicts of interest
There are no conflicts to declare.References
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- C. G. Palivan, R. Goers, A. Najer, X. Zhang, A. Car and W. Meier, Chem. Soc. Rev., 2016, 45, 377–411 RSC.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- S. F. M. van Dongen, M. Nallani, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem. – Eur. J., 2009, 15, 1107–1114 CrossRef CAS PubMed.
- J. Rodríguez-Hernández, F. Chécot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- S. Jain and F. S. Bates, Science, 2003, 300, 460–464 CrossRef CAS PubMed.
- J. A. Zupancich, F. S. Bates and M. A. Hillmyer, Macromolecules, 2006, 39, 4286–4288 CrossRef CAS.
- P. P. Ghoroghchian, G. Li, D. H. Levine, K. P. Davis, F. S. Bates, D. A. Hammer and M. J. Therien, Macromolecules, 2006, 39, 1673–1675 CrossRef CAS PubMed.
- L. Zhang and A. Eisenberg, Macromolecules, 1999, 32, 2239–2249 CrossRef CAS.
- E. V. Konishcheva, U. E. Zhumaev and W. P. Meier, Macromolecules, 2017, 50, 1512–1520 CrossRef CAS.
- C. Nardin, T. Hirt, J. Leukel and W. Meier, Langmuir, 2000, 16, 1035–1041 CrossRef CAS.
- M. S. Bretscher, Nature (London), New Biol., 1972, 236, 11 CrossRef CAS.
- P. F. Devaux, Biochemistry, 1991, 30, 1163–1173 CrossRef CAS PubMed.
- R. Stoenescu and W. Meier, Chem. Commun., 2002, 3016–3017 RSC.
- R. Stoenescu, A. Graff and W. Meier, Macromol. Biosci., 2004, 4, 930–935 CrossRef CAS PubMed.
- F. Liu and A. Eisenberg, J. Am. Chem. Soc., 2003, 125, 15059–15064 CrossRef CAS PubMed.
- A. F. Mason and P. Thordarson, ACS Macro Lett., 2016, 5, 1172–1175 CrossRef CAS.
- A. Wittemann, T. Azzam and A. Eisenberg, Langmuir, 2007, 23, 2224–2230 CrossRef CAS PubMed.
- G. Liu, S. Ma, S. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomaterials, 2010, 31, 7575–7585 CrossRef CAS PubMed.
- Q. Liu, J. Chen and J. Du, Biomacromolecules, 2014, 15, 3072–3082 CrossRef CAS PubMed.
- J. N. Israelachvili, Intermolecular and Surface Forces, 1991 Search PubMed.
- S. Schrage, R. Sigel and H. Schlaad, Macromolecules, 2003, 36, 1417–1420 CrossRef CAS.
- O. Casse, A. Shkilnyy, J. Linders, C. Mayer, D. Häussinger, A. Völkel, A. F. Thünemann, R. Dimova, H. Cölfen, W. Meier, H. Schlaad and A. Taubert, Macromolecules, 2012, 45, 4772–4777 CrossRef CAS.
- O. Terreau, L. Luo and A. Eisenberg, Langmuir, 2003, 19, 5601–5607 CrossRef CAS.
- C. J. Mable, R. R. Gibson, S. Prevost, B. E. McKenzie, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 16098–16108 CrossRef CAS PubMed.
- A. Walther, C. Barner-Kowollik and A. H. E. Müller, Langmuir, 2010, 26, 12237–12246 CrossRef CAS PubMed.
- A. Sundararaman, T. Stephan and R. B. Grubbs, J. Am. Chem. Soc., 2008, 130, 12264–12265 CrossRef CAS PubMed.
- P. Bhargava, Y. Tu, J. X. Zheng, H. Xiong, R. P. Quirk and S. Z. D. Cheng, J. Am. Chem. Soc., 2007, 129, 1113–1121 CrossRef CAS PubMed.
- A. O. Moughton, J. P. Patterson and R. K. O'Reilly, Chem. Commun., 2011, 47, 355–357 RSC.
- I. LaRue, M. Adam, M. Pitsikalis, N. Hadjichristidis, M. Rubinstein and S. S. Sheiko, Macromolecules, 2006, 39, 309–314 CrossRef CAS.
- S. Abbas, Z. Li, H. Hassan and T. P. Lodge, Macromolecules, 2007, 40, 4048–4052 CrossRef CAS.
- Q. Chen, H. Schönherr and G. J. Vancso, Small, 2010, 6, 2762–2768 CrossRef CAS PubMed.
- C. Maiti, R. Banerjee, S. Maiti and D. Dhara, Langmuir, 2015, 31, 32–41 CrossRef CAS PubMed.
- J. R. Lovett, N. J. Warren, S. P. Armes, M. J. Smallridge and R. B. Cracknell, Macromolecules, 2016, 49, 1016–1025 CrossRef CAS PubMed.
- M. Lee, S.-J. Lee and L.-H. Jiang, J. Am. Chem. Soc., 2004, 126, 12724–12725 CrossRef CAS PubMed.
- T.-H. Ku, M.-P. Chien, M. P. Thompson, R. S. Sinkovits, N. H. Olson, T. S. Baker and N. C. Gianneschi, J. Am. Chem. Soc., 2011, 133, 8392–8395 CrossRef CAS PubMed.
- X. Ji, H. Wang, Y. Li, D. Xia, H. Li, G. Tang, J. L. Sessler and F. Huang, Chem. Sci., 2016, 7, 6006–6014 RSC.
- M. C. M. van Oers, F. P. J. T. Rutjes and J. C. M. van Hest, J. Am. Chem. Soc., 2013, 135, 16308–16311 CrossRef CAS PubMed.
- N. J. W. Penfold, J. R. Lovett, P. Verstraete, J. Smets and S. P. Armes, Polym. Chem., 2017, 8, 272–282 RSC.
- R. Deng, M. J. Derry, C. J. Mable, Y. Ning and S. P. Armes, J. Am. Chem. Soc., 2017, 139, 7616–7623 CrossRef CAS PubMed.
- M. R. Molla, P. Prasad and S. Thayumanavan, J. Am. Chem. Soc., 2015, 137, 7286–7289 CrossRef CAS PubMed.
- E. Konishcheva, D. Häussinger, S. Lörcher and W. Meier, Eur. Polym. J., 2016, 83, 300–310 CrossRef CAS.
- O. Sedlacek, B. D. Monnery, S. K. Filippov, R. Hoogenboom and M. Hruby, Macromol. Rapid Commun., 2012, 33, 1648–1662 CrossRef CAS PubMed.
- M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- R. Hoogenboom, B. C. Moore and U. S. Schubert, Chem. Commun., 2006, 4010–4012 RSC.
- J. Ries and P. Schwille, BioEssays, 2012, 34, 361–368 CrossRef PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed.
- K. Bacia, S. A. Kim and P. Schwille, Nat. Methods, 2006, 3, 83 CrossRef CAS PubMed.
- V. Buschmann, B. Krämer, F. Koberling, R. Macdonald and S. Rättinger, Application Note PicoQuant, GmbH, Berlin, 2009 Search PubMed.
- E. V. Konishcheva, U. E. Zhumaev, M. Kratt, V. Oehri and W. Meier, Macromolecules, 2017, 50, 7155–7168 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization of polymers, 1H NMR of ABC-biotin, LSM of polymersomes formed by ABC and biotinylated polymers, FCS of ABC labeled with Bodipy 630/650, calculation of polymersome volume, and LSM of cloud-like aggregates. See DOI: 10.1039/c8cc09659g |
| This journal is © The Royal Society of Chemistry 2019 |