
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced photoelectrochemical hydrogen generation in neutral electrolyte using non-vacuum processed CIGS photocathodes with an earth-abundant cobalt sulfide catalyst†
Mingqing
Wang‡
 a,
Yung-Shan
Chang‡
b,
Chun-Wen
Tsao
b,
Mei-Jing
Fang
b,
Yung-Jung
Hsu
*bc and
Kwang-Leong
Choy
*a
a,
Yung-Shan
Chang‡
b,
Chun-Wen
Tsao
b,
Mei-Jing
Fang
b,
Yung-Jung
Hsu
*bc and
Kwang-Leong
Choy
*a
aUCL Institute for Materials Discovery, University College London, Roberts Building, Malet Place, London, WC1E 7JE, UK. E-mail: k.choy@ucl.ac.uk
bDepartment of Materials Science and Engineering, National Chiao Tung University, Hsinchu 30010, Taiwan. E-mail: yhsu@cc.nctu.edu.tw
cCenter for Emergent Functional Matter Science, National Chiao Tung University, Hsinchu 30010, Taiwan
First published on 31st January 2019
Abstract
This work reports the novelty of using eco-friendly and cost-effective non-vacuum Electrostatic Spray-Assisted Vapour Deposited Cu(In,Ga)SSe (CIGS) thin films as photocathodes, combined with the earth abundant cobalt sulfide (Co–S) as a catalyst to accelerate the kinetics of photogenerated electron transfer and hydrogen generation for photoelectrochemical water splitting. CdS and ZnO layers were subsequently deposited on top of the selenised CIGS films to increase the charge separation and lower the charge recombination for the photocathodes. In order to improve the lifetime and scalability of the CIGS photocathode and the other cell components, a photoelectrochemical test was conducted in a neutral electrolyte of 0.5 M Na2SO4 under simulated sunlight (AM 1.5G). Both the photocurrent densities and the onset potentials of the photocathodes were significantly improved by the electrodeposition of the low cost and earth-abundant Co–S catalyst, with a photocurrent density as high as 19.1 mA cm−2 at −0.34 V vs. reversible hydrogen electrode (RHE), comparable with and even higher than that of the control photocathode using rare and precious Pt as a catalyst.
With the efficiency achievement and progress made by academics and the photovoltaic industry, the cost of solar cells has been greatly lowered and become competitive with the price of conventional fuels. However, due to the intermittency and unpredictable properties of solar energy, it is becoming more relevant and useful to explore the development of an integrated device that combines both energy harvesting and energy storage. Photoelectrochemical (PEC) water splitting is an efficient way to absorb sunlight and transform the produced energy into hydrogen (H2), which can be easily stored as fuel and then oxidized by burning in air or in a fuel cell to release energy when required.
For direct PEC water splitting, the minimum theoretical electrical potential that is required to split water under standard conditions is 1.23 V (vs. standard hydrogen electrode, SHE). Owing to several factors such as cell resistance, irreversible processes etc., the actual voltage required to achieve water electrolysis is typically in the range of 1.8–2.0 V. Metal oxides have been widely studied as photoelectrodes in PEC water splitting due to their suitable band edge position and good chemical stability. For example, doped TiO2, Fe2O3, ZnO, BiVO4 and WO3 are generally considered as promising photoanode materials, while Cu2O, NiOx and Co3O4 are widely studied as photocathode materials.1 However, the band gap of most metal oxides is relatively too large to absorb sufficient sunlight, which limits the efficient absorption of visible light. As compared with metal oxides, CIGS has a tunable bandgap in the range of 1.0–2.4 eV, and its excellent light harvesting characteristics (i.e. absorption coefficients of 105 cm−1) allow a high light harvesting efficiency of close to unity. Solar cells using a CIGS absorber have achieved efficiency approaching 22.6%.2 Based on the research achievement on energy generation and urgent needs for energy storage, p-type CIGS semiconductor film has become a promising photocathode for PEC water splitting.3 The use of vacuum-based fabrication methods, such as multi-stage co-evaporation/sputtering processes for depositing CIGS absorbers, is very costly because of expensive instrumentation and the use of sophisticated vacuum systems. Therefore, significant effort has been invested in preparing CIGS films using non-vacuum approaches including spin-coating,4 hydrazine,5 quantum dots6 and electrodeposition.7 In our group, an Electrostatic Spray-Assisted Vapour deposition (ESAVD) approach using environmentally friendly precursor materials has been developed to deposit CIGS films.8 A thin layer of CdS followed by another thin layer of ZnO were deposited onto the CIGS layer to form a p–n junction and to avoid short circuiting, respectively, which further enhanced the electron transport for H2 production. Fig. 1 shows the device structure and energy level alignment in the CIGS based photocathodes. The band alignment in the photocathodes occurs in such a way that the minimum position of the conduction band decreases gradually from CIGS to CdS to ZnO, which accelerates the electron transfer from CIGS to ZnO. The high potential barrier of ZnO prevents the back flowing of electrons and decreases the charge recombination rate. After light absorption, the electrons and holes generated in CIGS are quickly separated at the CIGS/CdS p–n junction; holes would flow to the counter electrode through Mo and the external circuit, where oxygen (O2) is produced. While electrons flow from CIGS through CdS to the surface of ZnO, where H+ ions in the electrolyte are reduced to H2. A hydrogen evolution catalyst can promote the PEC water splitting efficiency by accelerating the H2 generation process. Platinum (Pt) has been normally used as a catalyst to improve the hydrogen evolution kinetics of CIGS/CIS photocathodes.3,9 However, Pt is very expensive and is scare in the earth, making it not suitable for large-scale application. Cobalt sulfide (Co–S) is an earth-abundant, cheap, and environmentally green material which can be processed at low temperature. In recent years, Co–S has been studied as a catalyst for H2 and O2 evolutions for TiO2, ZnO, and Si by virtue of its intriguing properties like good chemical/electrochemical stability and low free energy for hydrogen adsorption.10 Therefore, Co–S was selected as the alternative catalyst to replace Pt for the CIGS photocathode in this work.
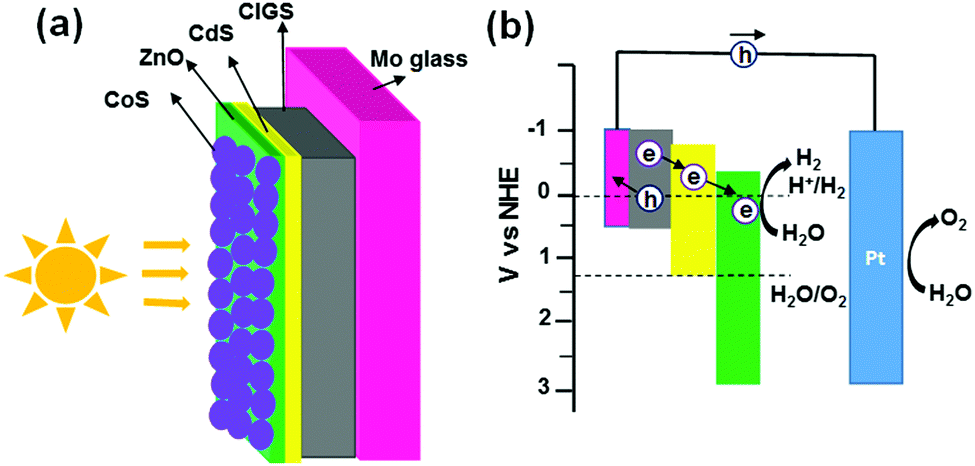 | ||
| Fig. 1 (a) Device structure and (b) energy alignment of charge transfer for CIGS-based photocathodes toward PEC water splitting. | ||
Uniform CIGS thin films have been deposited by the non-vacuum ESAVD method followed by selenization at 550 °C for 30 min. Fig. 2 shows the surface morphology and purity of the CIGS absorber characterized using SEM, XRD and Raman analysis. The SEM images in Fig. 2a and b show the CIGS absorber composed of large grains with grain size circa 1 micrometer in size. XRD and Raman results of Fig. 2c and d illustrate that the deposited absorber is well crystallized without any other undesirable binary or ternary impurities.
 | ||
| Fig. 2 Characterization of the CIGS absorber: (a) surface morphology; (b) cross-sectional SEM image, (c) XRD pattern and (d) Raman spectrum. | ||
In order to make the PEC results easier for analysis and comparison, two electrochemical potential definitions are adopted in the following work. The photocurrents measured relative to the SCE reference electrode were converted to photocurrents vs. a reversible hydrogen electrode (RHE) using the following equation ERHE = ESCE + 0.059pH + 0.244. On the other hand, the onset potential (Vonset) is defined as the potential reaching a cathodic photocurrent density of 0.1 mA cm−2 and is used as another key parameter to assess the PEC performance of the photocathodes.
CIGS is a p-type semiconductor with strong light absorption characteristics. If bare CIGS was used as a PEC photocathode, it would not perform well due to the lack of favorable energetics alignment with the electrolyte. Thus, this would result in inefficient separation of photogenerated charge carriers. As Fig. S1 (ESI†) shows, the photocurrent generation of pure CIGS was very low, less than 1.0 mA cm−2 even when a large bias was exerted. Whereas, the deposition of a thin layer of CdS on top of CIGS could form a solid-state p–n junction and lead to more efficient charge separation.11 As compared with CdS, ZnO has a wider bandgap (3.37 eV) which is more favorable for driving the water reduction reaction while maintaining suitable valence and conduction band positions. Thus, by depositing another thin layer of ZnO on top of CdS, it would form a better alignment between the ZnO/CdS/CIGS interfaces, which would help to drive the charge transport further and minimize charge recombination. Fig. 3 shows J–V curves obtained from CIGS/CdS and CIGS/CdS/ZnO photocathodes in 0.5 M aqueous Na2SO4 solution (pH = 7) under continuous and chopped simulated sunlight (AM1.5G) illuminations. Both of the photocathodes demonstrated very low and negligible dark current between −0.35 and +1.0 V vs. RHE, suggesting that the observed photocurrents were mainly from the reduction of H2O to H2. Under illumination, at −0.35 V vs. RHE, the photocurrent density of the CIGS/CdS/i-ZnO photocathode was greatly improved to 6.46 mA cm−2 compared with 2.78 mA cm−2 for the CIGS/CdS photocathode. Furthermore, the photocurrent onset potential of the CIGS/CdS/i-ZnO photocathode was anodically shifted from +0.65 V vs. RHE of CIGS/CdS to +0.69 V vs. RHE, implying efficient charge transfer and lower charge recombination. The increase in the photocurrent and the anodic shift of the photocurrent onset potential after ZnO layer deposition are directly associated with the high potential barrier of ZnO as shown in Fig. 1, which helps to prevent the back flowing of electrons, thus decreasing the charge recombination and increasing the photocurrent.
 | ||
| Fig. 3 J–V curves for CIGS photocathodes with different modification layers under (a) continuous and (b) chopped AM 1.5G sunlight illumination. | ||
For the commercialization of PEC systems at the terawatt scale, devices should ideally reach or exceed 10% solar-to-hydrogen (STH) conversion efficiencies. The PEC water splitting system works the best at either a very high or very low pH value, where the concentration of H+/OH− is the greatest. However, most of the semiconductor materials with strong light absorption such as Si, GaN, and CIGS degrade rapidly under extreme pH values. Under neutral pH, a hydrogen evolution reaction (HER) catalyst would need to be used to promote the water splitting kinetics. Conventionally, Pt is used as the catalyst. However, Pt is very expensive. Thus, earth-abundant Co–S was selected as the HER catalyst in this work and electrodeposited onto the CIGS photocathodes. A combination of SEM and XRD was used to characterize the surface morphology, composition and crystal structure of the CIGS photocathode with electrodeposited Co–S catalyst and the results are presented in Fig. 4. The SEM image (Fig. 4a) reveals a thin layer of Co–S (from energy-dispersive X-ray spectroscopy in Fig. 4c) covering the top surface of the photocathode. From the XRD pattern (Fig. 4b) of the photocathode, no obvious peaks of Co–S were observed. This demonstrates that the deposited Co–S layer was amorphous. Fig. 4d further shows that the CIGS/CdS/i-ZnO/Co–S photocathode has strong absorption in the visible range, which is beneficial for harvesting solar energy to promote solar H2 production. Note that pure Co–S exhibited considerably weak visible absorption (Fig. S2, ESI†), suggesting that most of the photons were harvested by the underneath CIGS, CdS and ZnO, and Co–S simply functioned as the HER catalyst.
 | ||
| Fig. 4 (a) SEM image, (b) XRD pattern, (c) SEM-EDX analysis, and (d) UV-vis absorption spectrum for the CIGS/CdS/i-ZnO/Co–S photocathode. In (b) and (d), the results of the Mo substrate were also included for comparison. | ||
Fig. 5 shows the J–V curves for CIGS/CdS/ZnO photocathodes coated with Pt or Co–S as the catalyst under both continuous and chopped illumination. After the deposition of the HER catalysts, the onset potential of the photocathodes anodically shifted from +0.69 to +0.85 V vs. RHE for Pt and to +0.89 V vs. RHE for Co–S, respectively. As can be seen from Fig. 5a and b, the photocurrent density (Jph) was also greatly increased after deposition of both catalysts. When Co–S was used as a catalyst, the Jph of the CIGS photocathode reached 19.1 mA cm−2 at −0.34 V vs. RHE, which is even higher than that of the photocathode with the traditional Pt catalyst (Jph of 18.20 mA cm−2). The STH efficiency was further estimated from the expression η = [Jph × (1.23 − |Vbias|)]/Plight, where Jph is the photocurrent density at the measured potential, Plight is the power density of incident light (100 mW cm−2), and Vbias is the applied potential.12 At a moderate potential (0 V vs. RHE), the CIGS/CdS/ZnO/CoS photocathode achieved an STH efficiency of 2.5%. Although this value was less than those attained from the state-of-the-art photocathodes, the current work still posed great impact considering that the performance enhancement by Co–S deposition was significant. The anodic shift of the onset potential of the photocathode and the increase of photocurrent density after deposition of the Co–S catalyst are noteworthy, reflecting the accelerated and efficient transfer of photo-induced carriers from CIGS to react with H+/H2 redox couples in the electrolyte.
 | ||
| Fig. 5 J–V curves under (a) continuous and (b) chopped illumination for CIGS/CdS/i-ZnO photocathodes with Pt and Co–S as catalysts. | ||
In summary, non-vacuum ESAVD has been successfully used to deposit CIGS as photocathodes. The PEC water splitting performance of the ESAVD deposited photocathodes in a neutral solution has been investigated. The lower cost and earth-abundant Co–S was applied as a HER catalyst for H2 production. The integrated device produced photocurrent as high as 19.1 mA cm−2 at −0.34 V vs. RHE, which is comparable with and even better than the control photocathode using expensive Pt as a catalyst. This work has demonstrated the potential of combining the earth-abundant catalyst with widely studied chalcogenide absorber for solar water splitting. In addition, the PEC test in our work was conducted in neutral electrolyte, which can help to avoid the degradation of the cell components (absorber, ZnO, gaskets, connections, and membranes) and provide added advantages for the future scale-up of the technique. The composition and bandgap of the chalcogenide absorber could be further optimized to promote the light absorption and driving force for water splitting to achieve better PEC efficiency.
The authors acknowledge the financial support of Innovate UK under High-Prospect project (102470). This work was also financially supported by the Ministry of Science and Technology (MOST) of Taiwan under grant MOST 107-2113-M-009-004. Y.-J. Hsu also acknowledges the budget support from the Center for Emergent Functional Matter Science of National Chiao Tung University from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education in Taiwan.
Conflicts of interest
There are no conflicts to declare.References
- H. Pan, Renewable Sustainable Energy Rev., 2016, 57, 584–601 CrossRef CAS.
- P. Jackson, R. Wuerz, D. Hariskos, E. Lotter, W. Witte and M. Powalla, Phys. Status Solidi RRL, 2016, 10, 583–586 CrossRef CAS.
- (a) D. Yokoyama, T. Minegishi, K. Maeda, M. Katayama and J. Kubota, et al. , Electrochem. Commun., 2010, 12, 851–853 CrossRef CAS; (b) M. G. Mali, H. Yoon, B. N. Joshi, H. Park, S. S. Al-Deyab, D. C. Lim, S. Ahn, C. Nervi and S. S. Yoon, ACS Appl. Mater. Interfaces, 2015, 7, 21619–21625 CrossRef CAS PubMed; (c) A. Azarpira, M. Lublow, A. Steigert, P. Bogdanoff and D. Greiner, et al. , Adv. Energy Mater., 2015, 5, 1402148 CrossRef; (d) N. Guijarro, M. S. Prevot, X. Y. Yu, X. A. Jeanbourquin, P. Bornoz, W. Bouree, M. Johnson, F. Le Formal and K. Sivula, Adv. Energy Mater., 2016, 6, 1501949 CrossRef.
- A. R. Uhl, C. Fella, A. Chirila, M. R. Kaelin, L. Karvonen, A. Weidenkaff, C. N. Borca, D. Grolimund, Y. E. Romanyuk and A. N. Tiwari, Prog. Photovoltaics, 2012, 20, 526–533 CAS.
- T. K. Todorov, O. Gunawan, T. Gokmen and D. B. Mitzi, et al. , Thin Solid Films, 2000, 361, 396–399 Search PubMed.
- S. M. McLeod, C. J. Hages, N. J. Carter and R. Agrawal, Prog. Photovoltaics, 2015, 23, 1550–1556 CAS.
- R. N. Bhattacharya, J. F. Hiltner, W. Batchelor, M. A. Contreras, R. N. Noufi and J. R. Sites, Thin Solid Films, 2000, 361, 396–399 CrossRef.
- (a) M. A. Hossain, M. Q. Wang and K. L. Choy, ACS Appl. Mater. Interfaces, 2015, 7, 22497–22503 CrossRef CAS PubMed; (b) M. Q. Wang, X. H. Hou, J. P. Liu, K. Choy, P. Gibson, E. Salem, D. Koutsogeorgis and W. Cranton, Phys. Status Solidi A, 2015, 212, 72–75 CrossRef CAS; (c) K. L. Choy, Prog. Mater. Sci., 2003, 48, 57–170 CrossRef CAS.
- (a) B. Koo, S. W. Nam, R. Haight, S. Kim and S. Oh, et al. , ACS Appl. Mater. Interfaces, 2017, 9, 5279–5287 CrossRef CAS PubMed; (b) W. Septina, Gunawan, S. Ikeda, T. Harada, M. Higashi, R. Abe and M. Matsumura, J. Phys. Chem. C, 2015, 119, 8576–8583 CrossRef CAS; (c) R. C. Valderrama, P. J. Sebastian, J. P. Enriquez and S. A. Gamboa, Sol. Energy Mater. Sol. Cells, 2005, 88, 145–155 CrossRef CAS; (d) W. Septina, M. Sugimoto, D. Chao, Q. Shen, S. Nakatsuka, Y. Nose, T. Harada and S. Ikeda, Phys. Chem. Chem. Phys., 2017, 19, 12502–12508 RSC; (e) H. Kumagai, T. Minegishi, N. Sato, T. Yamada, J. Kubota and K. Domen, J. Mater. Chem. A, 2015, 3, 8300–8307 RSC.
- (a) Y. Chen, P. D. Tran, P. Boix, Y. Ren, S. Y. Chiam, Z. Li, K. W. Fu, L. H. Wong and J. Barber, ACS Nano, 2015, 9, 3829–3836 CrossRef CAS PubMed; (b) M. Q. Wang, M. A. Hossain and K. L. Choy, Sci. Rep., 2017, 7, 22109 Search PubMed; (c) Y. J. Sun, C. Liu, D. C. Grauer, J. K. Yano, J. R. Long, P. D. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 17699–17702 CrossRef CAS PubMed.
- T. J. Jacobsson, C. Platzer-Bjorkman, M. Edoff and T. Edvinsson, Int. J. Hydrogen Energy, 2013, 38, 15027–15035 CrossRef CAS.
- (a) J.-M. Li, H.-Y. Cheng, Y.-H. Chiu and Y.-J. Hsu, Nanoscale, 2016, 8, 15720–15729 RSC; (b) Y.-H. Chiu, T.-H. Lai, C.-Y. Chen, P.-Y. Hsieh, K. Ozasa, M. Niinomi, K. Okada, T.-F. M. Chang, N. Matsushita, M. Sone and Y.-J. Hsu, ACS Appl. Mater. Interfaces, 2018, 10, 22997–23008 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section and supplementary figures. See DOI: 10.1039/c8cc09426h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |