DOI:
10.1039/C8CC09077G
(Feature Article)
Chem. Commun., 2019,
55, 1859-1878
Recent development on the [5+2] cycloadditions and their application in natural product synthesis
Received
15th November 2018
, Accepted 15th January 2019
First published on 16th January 2019
Abstract
The [5+2] cycloaddition is one kind of fascinating and powerful reaction to access seven-membered ring systems existing in complex natural products and pharmaceuticals. In this review, recent developments on the [5+2] cycloadditions and their application in the synthesis of complex natural products are discussed. A focus is given to the way in which the [5+2] cycloadditions have influenced the logical process of the synthesis. This article is organized by reaction types, aiming to provide a clear clue to the latest research trends.

Kai Gao
| Kai Gao received his PhD from Dalian Institute of Chemical Physics, CAS in 2012. From 2016 to 2018, he was a postdoctoral fellow working with Prof. Wei-Min Dai at Hong Kong University of Science and Technology (HKUST). In 2018, he joined the Institute of Medicinal Natural Product, School of Advanced Study at Taizhou University as a lecturer. His current research interests focus on the development of hypervalent iodine-mediated reactions and their application in natural products synthesis. |

Yong-Gang Zhang
| Yong-Gang Zhang received his PhD from University of Science and Technology of China in 2014. Since 2018, he joined the Institute of Medicinal Natural Product, School of Advanced Study at Taizhou University as a lecturer. His current research interests focus on metal-catalyzed synthetic methodologies and their application in natural products synthesis. |

Zhiming Wang
| Zhiming Wang received his PhD from Zhejiang University in 2007. From 2008 to 2010, he was a postdoctoral fellow working with Prof. Ohyun Kwon at University of California, Los Angeles (UCLA). He began his independent career as a professor at Changzhou University in September 2010. In 2018, he moved to Taizhou University. His current research interests focus on the development of transition-metal-catalyzed reaction for the synthesis of heterocycles and their applications in biochemistry. |

Hanfeng Ding
| Hanfeng Ding received his PhD from Zhejiang University in 2008. From 2009 to 2011, he was a postdoctoral fellow working with Dr David Y.-K. Chen in Prof. K. C. Nicolaou's group at CSL@Biopolis, Singapore. He began his independent career as a professor in the Department of Chemistry at Zhejiang University in September 2011. His current research interests focus on the development of novel strategies and methodologies for the total synthesis of structurally complex and biologically active natural products. |
1. Introduction
The development of efficient methodologies and the design of novel strategies for the rapid construction of molecular complexity in a stereoselective manner are of great significance in organic synthesis. With no doubt, cycloaddition reactions are among the most powerful chemical transformations that allow multiple carbon–carbon or carbon–heteroatom bonds to form in one single step, often with satisfactory regio- and stereoselectivities. In the past few decades, various types of these reactions, such as [2+2], [3+2] and [4+2] cycloadditions, have been well developed for the effective assembly of the corresponding four to six-membered carbocycles.1a,b In comparison, the synthesis of seven-membered rings through a cycloaddition pathway has been less explored. This process is largely driven by the entropic factors derived from the changing of the internal rotations/translations of chemical bonds in the course of cyclization (especially for the intramolecular reactions in larger rings), and also influenced by the nonbonding interactions in the transition state.1c,d Thus, direct cyclization is generally difficult without activation of the substrates.1e Recently, the continuing identification of biologically active natural products that contain a seven-membered skeleton in the core structure has stimulated considerable advancements toward their syntheses. Among them, the [5+2] cycloadditions turned out to be versatile methods, which played important roles in the total syntheses of related complex natural products.2
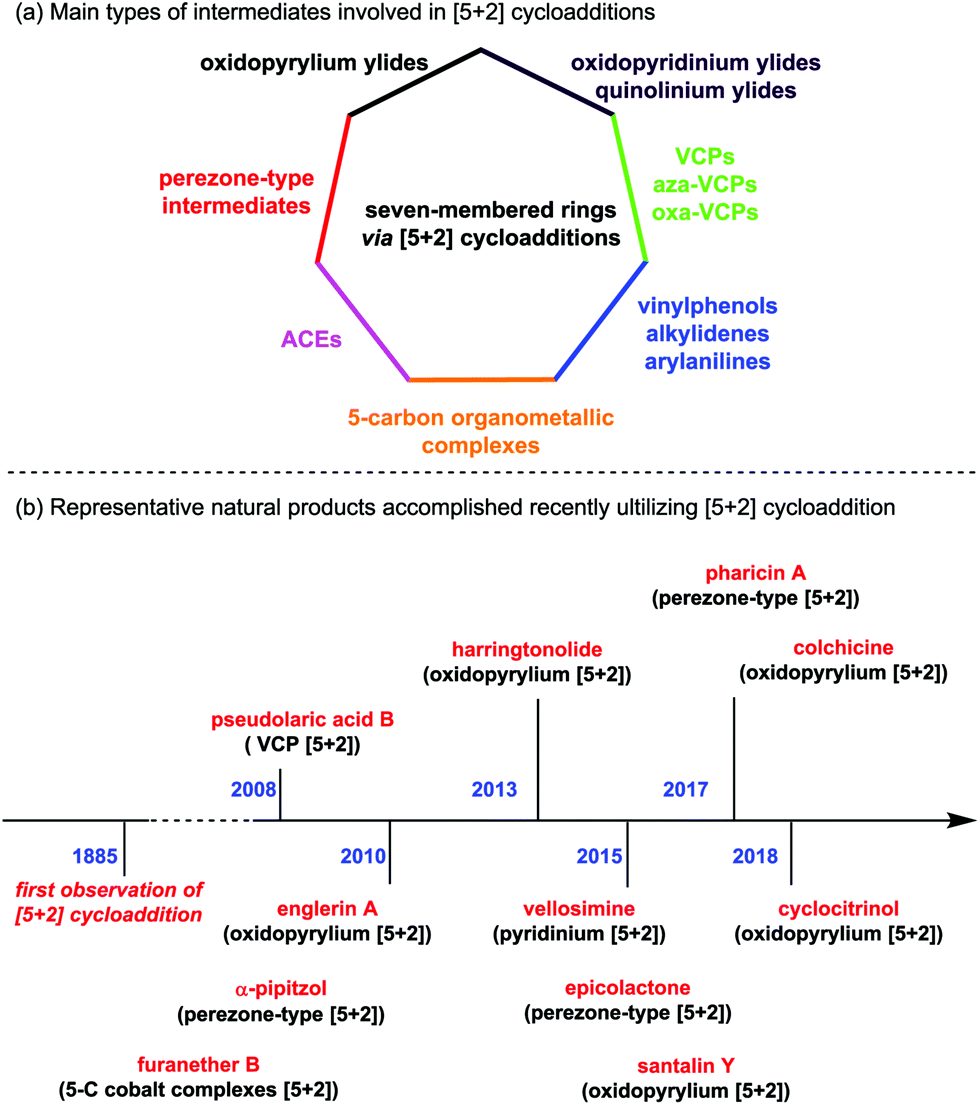
Since the first example described in 1885 by by Anschütz and Leather,3 [5+2] cycloadditions have been paid considerable attention. Apart from miscellaneous works on formal [5+2] cycloaddition4 and [5+2] photocycloaddition,5 to the best of our knowledge, there are mainly seven types of [5+2] cycloaddition reactions divided according to the structures of the corresponding precursors (Fig. 1a): (1) oxidopyrylium-mediated [5+2] cycloadditions; (2) oxidopyridinium- and quinolinium-mediated [5+2] cycloadditions; (3) perezone-type [5+2] cycloadditions; (4) [5+2] cycloadditions of vinyl substituted three-membered rings, including vinylcyclopropanes (VCPs), vinylaziridines (aza-VCPs) and vinyloxiranes (oxa-VCPs); (5) [5+2] cycloadditions of five-carbon organometallic complexes; (6) [5+2] cycloadditions of 3-acyloxy-1,4-enynes (ACEs); and (7) formal [5+2] cycloadditions of vinylphenols, alkylidenes and arylanilines. During the past decade, significant progress on this reaction has been made, especially in the field of natural product synthesis (see Fig. 1b for the representative examples as shown by the time axis).
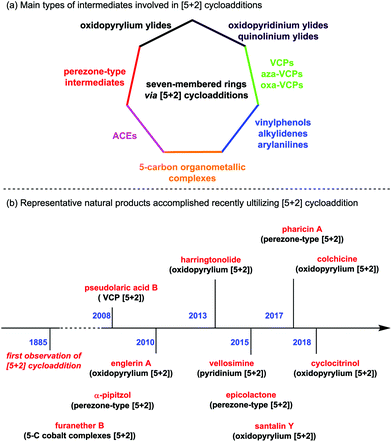 |
| | Fig. 1 (a) Main types of intermediates involved in [5+2] cycloadditions. (b) Representative natural products accomplished recently utilizing [5+2] cycloaddition as a key step. | |
A comprehensive review on the [5+2] cycloadditions was reported by Ylijoki and Stryker in 2012.6 On the other hand, Pellissier summarized the progress of [5+2] cycloadditions in two different time span,7 and the Li group delivered a review by highlighting the application of [5+2] cycloadditions in natural product synthesis.8 Very recently, Murelli et al. also disclosed their review devoted solely to oxidopyrylium cycloaddition chemistry.9 In view of the incessant springing up of new results in this hot research area, we feel it is very urgent to summarize and update the progress of [5+2] cycloaddition reactions and their corresponding application in natural product synthesis by carefully selected examples over the past decade. Due to the lack of application in natural product synthesis, the last two types of [5+2] cycloadditions mentioned above will not be discussed.10,11 We hope this feature article could serve as a meaningful and clear clue to those who are interested in [5+2] cycloaddition chemistry on the latest research trends.
2. Oxidopyrylium-mediated [5+2] cycloaddition
2.1 [5+2] cycloaddition of oxidopyrylium ylides from acetoxy-pyranones and 3-hydroxy-4-pyranones
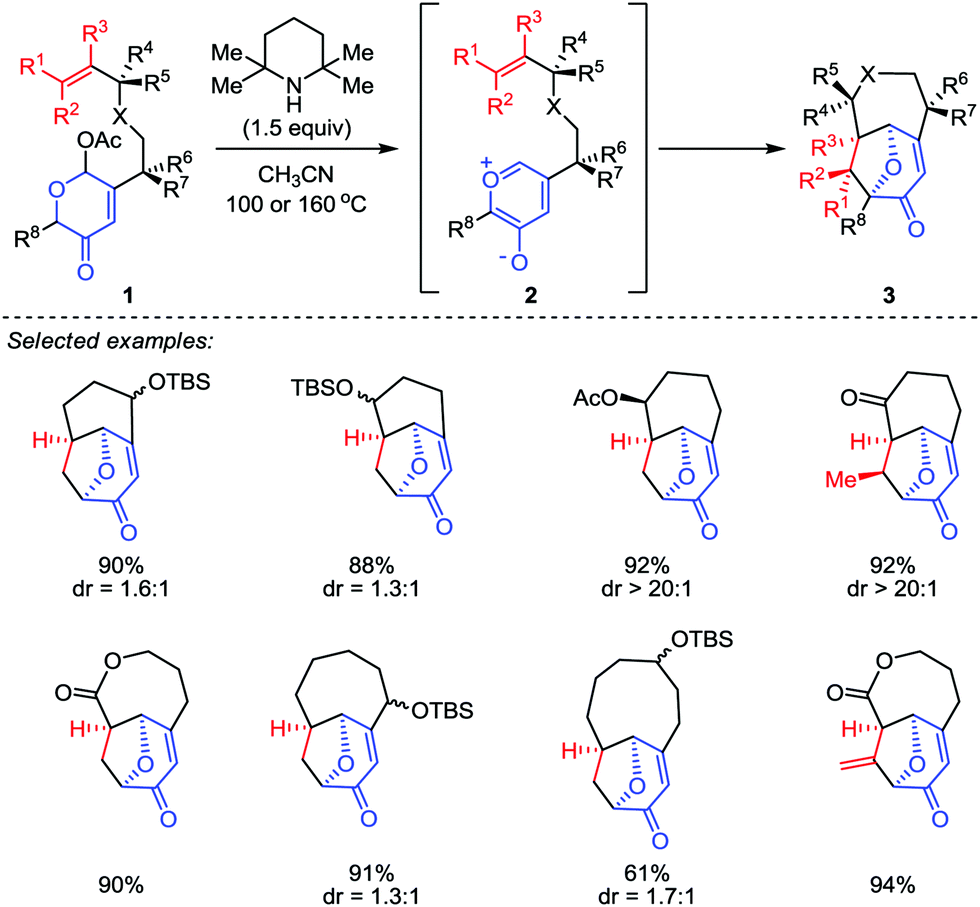
Although the construction of the bridged seven-membered ring systems has been extensively investigated,12 no general reactions were disclosed for the direct and efficient assembly of such motifs. Due to the importance of these bicyclic skeletons which are widely found in a number of biologically active natural products (such as ingenol and cyclocitrinol),13,14 their construction continues to capture the attention of synthetic chemists. It was not until 2015, a breakthrough was made by Li and co-workers.15 In their work, a type II intramolecular [5+2] cycloaddition was carried out by using a well-designed acetoxypyranones 1 with a dienophile tether at the C3 position as precursors (Scheme 1). Promoted by 2,2,6,6-tetramethyl-piperidine (TMP) in a sealed tube at elevated temperature (100 or 160 °C), a series of functionalized bridged bicyclic products 3, including bicyclo[4.3.1]decanes, bicyclo[4.4.1]undecanes, bicyclo[5.4.1]dodecanes and bicyclo[6.4.1]tridecanes, could be afforded in excellent yields.
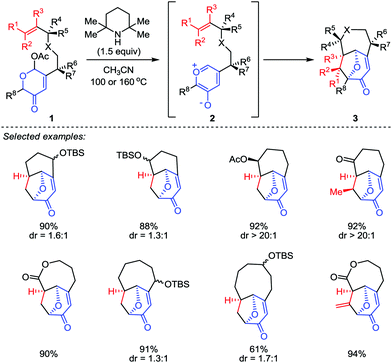 |
| | Scheme 1 Type II intramolecular [5+2] cycloaddition by Li and co-workers. | |
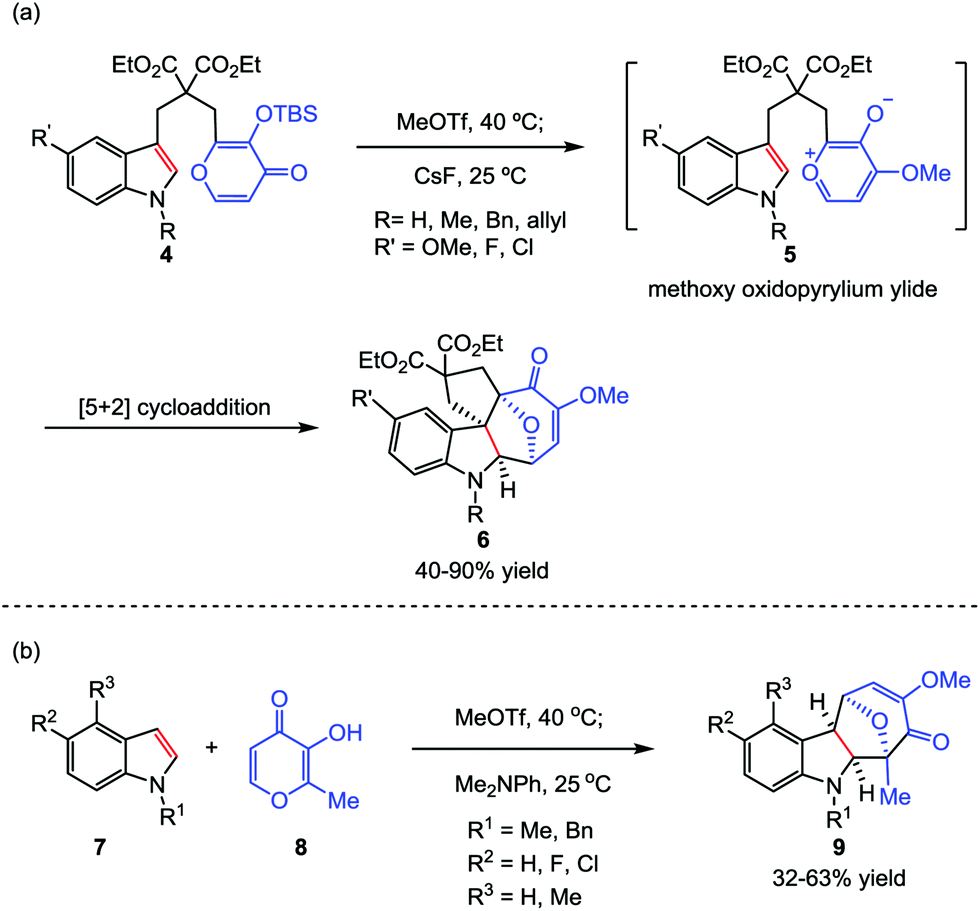
Considering that the high temperature which was required to generate the oxidopyrylium ylide would limit the overall scope and utility of this reaction, the Li group then sought inspiration from the group-transfer strategy developed by Wender and Mascareñas.16 To this end, MeOTf was chosen as a methylating reagent. Its reaction with kojic acids 4 at 40 °C successfully delivered the corresponding methoxy pyrylium salts. By treatment with CsF at room temperature, compound 4 was conveniently transformed to the oxidopyrylium ylides 5. This protocol enables both intra- and intermolecular oxidopyrylium ylide-indole [5+2] cycloadditions proceed smoothly, providing highly functionalized and stereochemically challenging oxa-cyclohepta[b]indoles 6 and 9 in moderate to high yields, respectively (Scheme 2).17
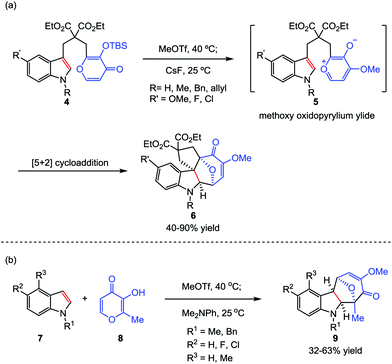 |
| | Scheme 2 Oxidopyrylium ylide-indole [5+2] cycloadditions by Li and co-workers. | |
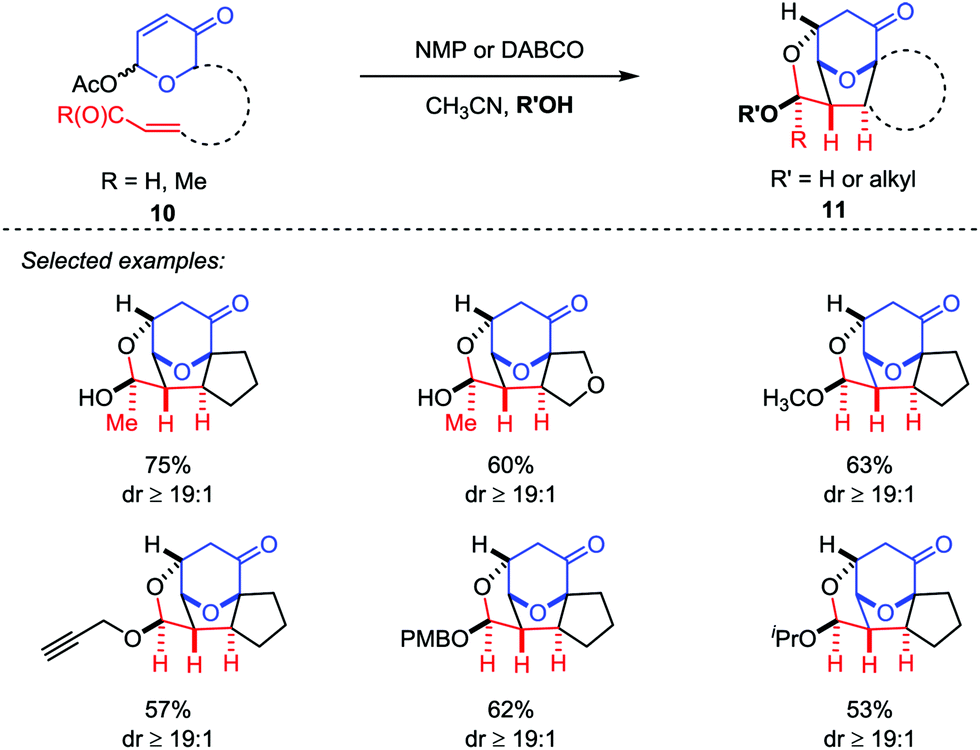
Recently, Tantillo and Mitchell described an oxidopyrylium-alkene [5+2] cycloaddition/oxa-Michael addition cascade sequences by the employment of acetoxypyranone-enals (or enones) 10 as oxidopyrylium ylide precursors with a variety of potential nucleophiles, which provided a platform to access the caged ethers 11 bearing up to four new bonds and six contiguous stereocenters with good yields and excellent diastereoselectivities (Scheme 3).18
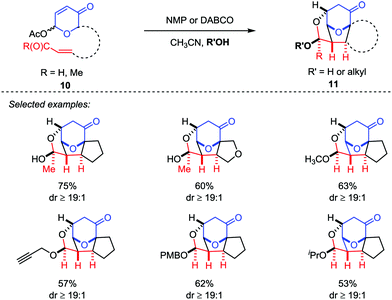 |
| | Scheme 3 Oxidopyrylium-alkene [5+2] cycloaddition/oxa-Michael addition cascade by Tantillo et al. | |
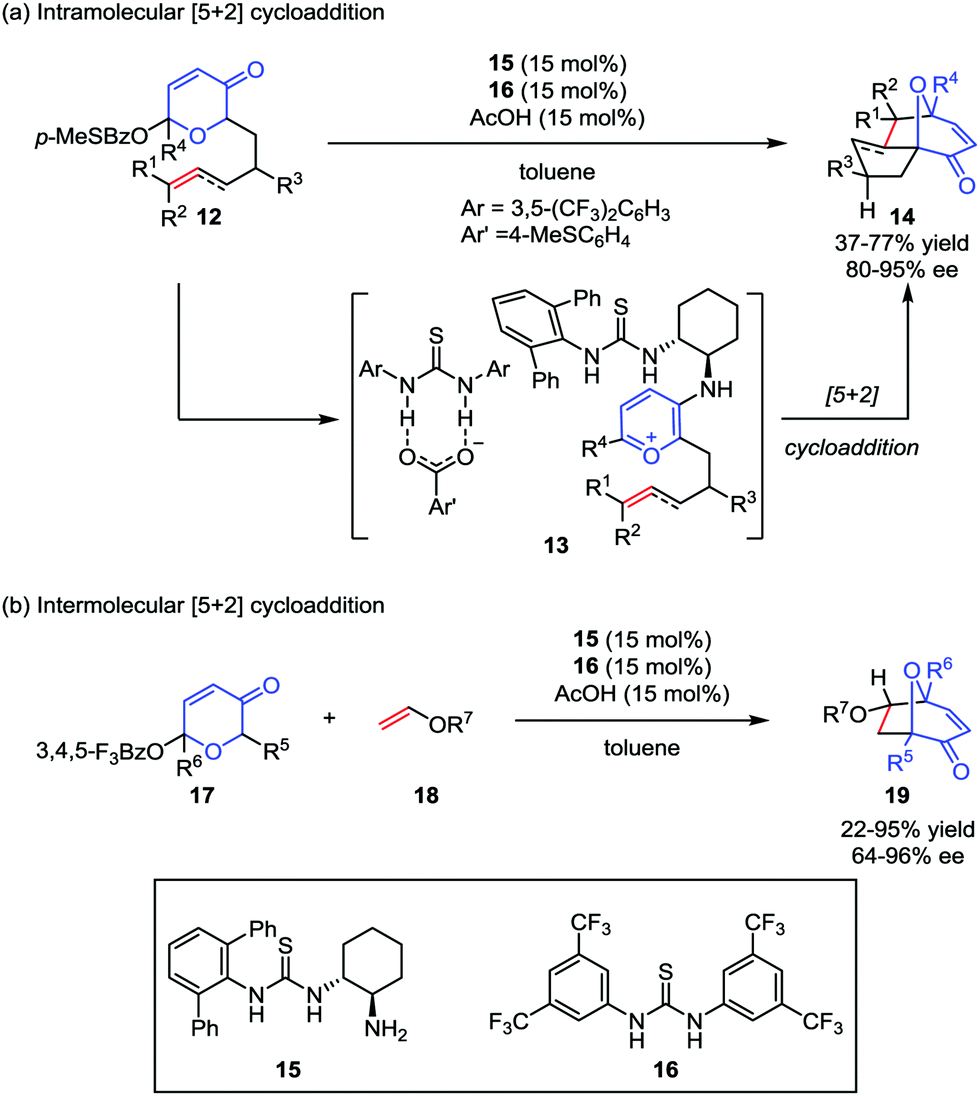
Despite the widespread use of oxidopyrylium-mediated [5+2] cycloaddition in organic synthesis, asymmetric examples, especially for enantioselective catalytic methods are rather limited. As one of their research interests in small-molecule chiral hydrogen-bond-donor catalysis, Jacobsen and co-workers envisioned the potential application of anion-binding catalysis in the oxidopyrylium formation and the following cycloaddition. Later, they developed a dual thiourea catalyst system consisting of a chiral primary aminothiourea 15 and another achiral thiourea 16 for intramolecular oxidopyrylium [5+2] cycloaddition of racemic acetoxypyranone 12, which became the first enantioselective approach to valuable tricyclic products 14 with up to 77% yield and 95% ee (Scheme 4a).19a It was found that the generation of thiourea-complexed aminopyrylium salts 13 was necessary to undergo cycloaddition with high enantioselectivities. Shortly after this work, they also success-fully achieved an intermolecular [5+2] cycloaddition reaction by using the electron-deficient pyranones 17 in the presence of the same dual thiourea catalyst system,19b providing the cycloadducts 19 in low to good yields and with moderate to excellent enantioselectivities (Scheme 4b).
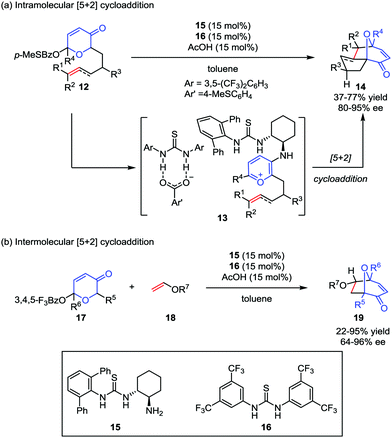 |
| | Scheme 4 The enantioselective oxidopyrylium-alkene [5+2] cycloaddition by Jacobsen and co-workers. | |
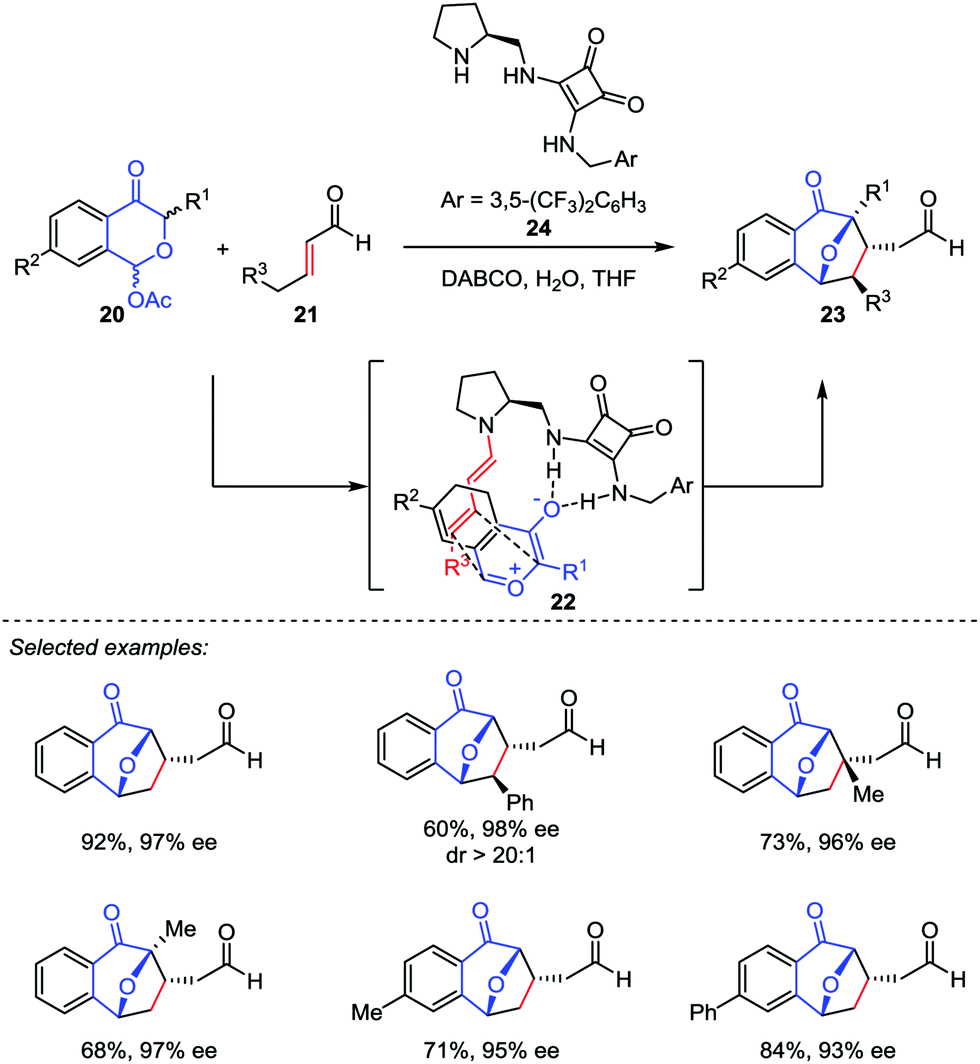
Based on the pioneering work of Jacobsen, Reyes and Vicario proposed another asymmetric catalytic model featured by dienamine activation.20 They reported a catalytic enantio-selective [5+2] cycloaddition between benzopyrylium ylides generated in situ from 1-acetoxyisochroman-4-ones 20 and α,β-unsaturated aldehydes 21 in the presence of a bifunctional secondary amine/squaramide catalyst 24 (Scheme 5). This method provides the direct access to 8-oxabicyclo[3.2.1]octane frameworks 23 with excellent dr and ee values. Mechanistically, the secondary amine catalyst 24 reacted with the enal 21 to form an activated dienamine intermediate 22, which played as an electron-rich alkene in this [5+2] cycloaddition. As a comparison, the chiral aminocatalyst 15 condensed with benzoyloxypyranone derivatives 12 to generate the reactive pyrylium ylide 13 in Jacobsen's process (Scheme 4).19
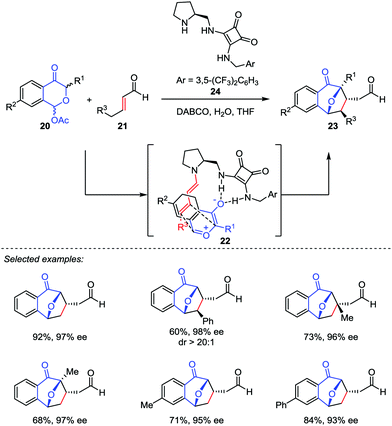 |
| | Scheme 5 The enantioselective oxidopyrylium-alkene [5+2] cycloaddition by Vicario and co-workers. | |
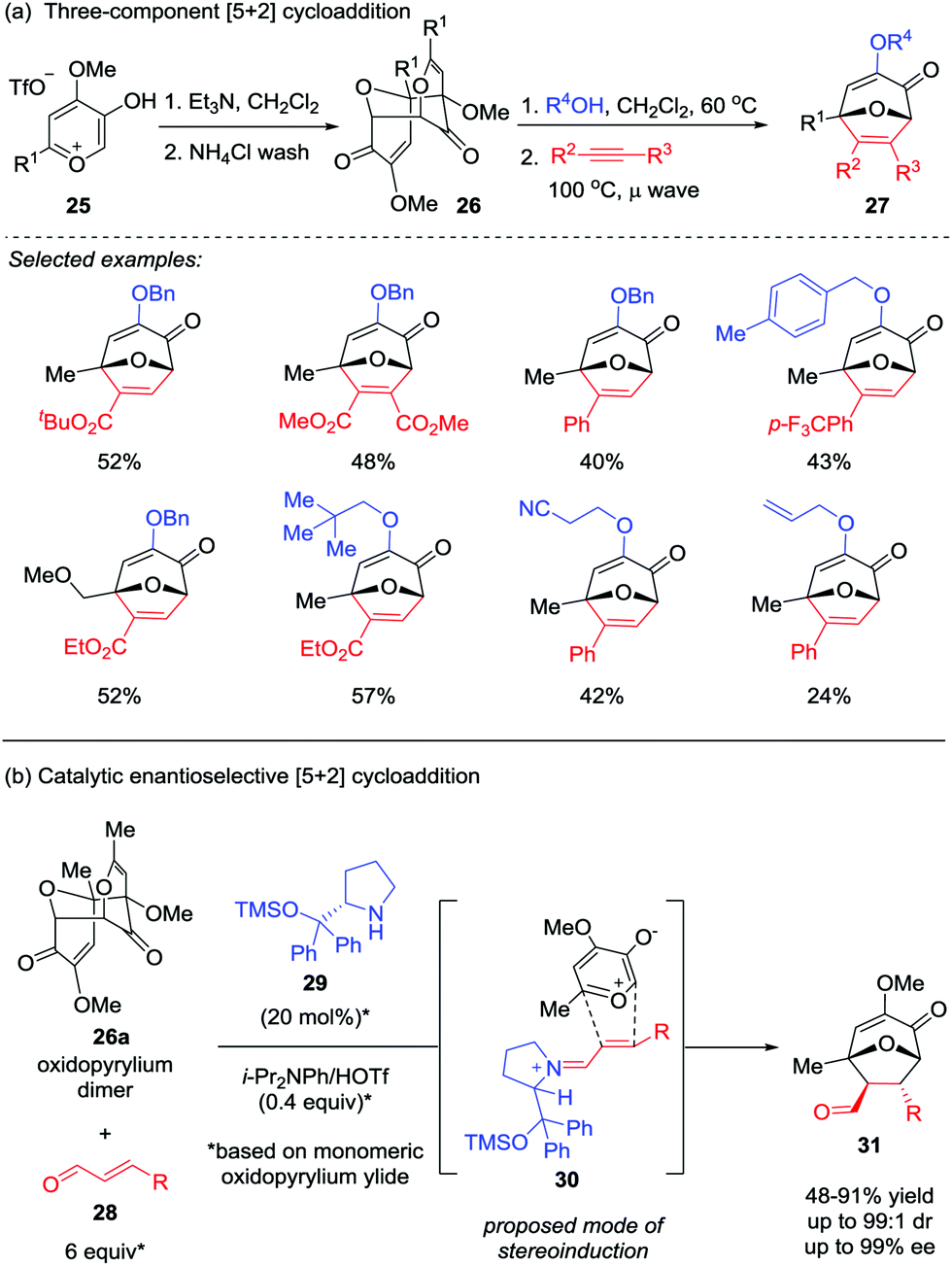
In most cases, the facile tautomerization or rearrangement of α-hydroxy-γ-pyrones to generate the oxidopyrylium ylides that are capable of undergoing [5+2] cycloaddition is only effective in an intramolecular fashion, presumably due to the short lifespan of the reactive ylide.21 To overcome this limitation, the Wender group firstly employed a methyl triflate-derived pre-ylide salt in a modified version of this reaction in 1991.16 Recently, Murelli and Brenner-Moyer prepared an oxidopyrylium ylide dimer 26 derived from methyl triflate salt 25, which was then subjected to the three-component [5+2] cycloaddition cascade reaction with an alcohol and an alkyne (Scheme 6a).22a By slightly heating a mixture of the purified oxidopyrylium dimer 26 in the presence of the alcohol prior to the addition of alkyne, a broad range of oxabicyclic compounds 27 were obtained in moderate yields. Interestingly, oxidopyrylium dimer 26a could also be employed as ylide source in an asymmetric version (Scheme 6b),22b enabling the first catalytic enantioselective [5+2] cycloaddition of a 3-hydroxy-4-pyrone-derived oxidopyrylium ylide in the presence of catalyst 29 through chiral iminium activation of β-substituted enal 28. Of note, the products 31 possess four stereocenters were formed with up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and 99% ee.
:1 dr and 99% ee.
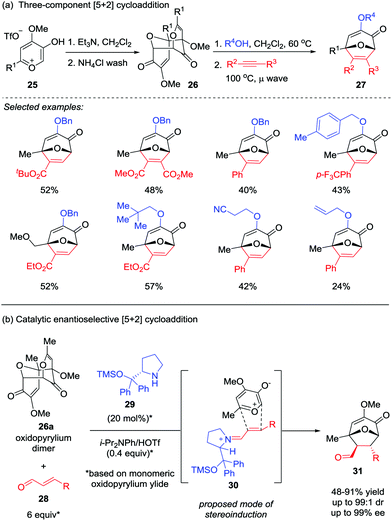 |
| | Scheme 6 [5+2] Cycloaddition of oxidopyrylium dimer 26 by Murelli and Brenner-Moyer. | |
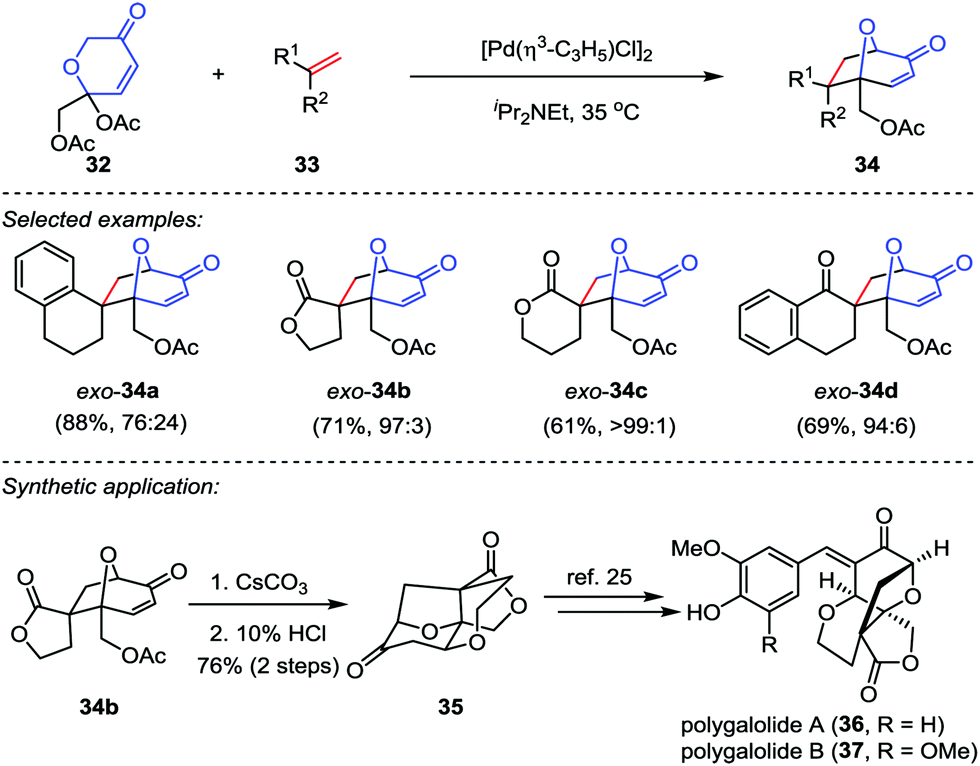
Recently, Suga and co-workers described an efficient methodology to generate oxidopyrylium ylides from 2H-pyran-3(6H)-ones 32 through [Pd(η3-C3H5)Cl]2 catalysis,23 which subsequently underwent the [5+2] cycloaddition with a variety of dipolarophiles 33 to afford the cycloadducts 34 in good to excellent yields (Scheme 7). To demonstrate the synthetic utility of this reaction, the cycloadduct 34b was converted to tetracycle 35 through a saponification/intramolecular oxa-Michael addition/lactonization sequence.24 Following the reported procedures from Nakamura and Hashimoto,25 polygalolides A (36) and B (37) could be uneventfully accessed.
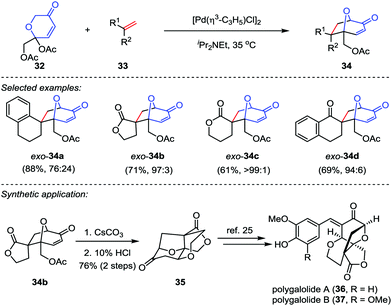 |
| | Scheme 7 The [Pd(η3-C3H5)Cl]2-catalyzed [5+2] cycloaddition by Suga and co-workers. | |
2.2 Natural product synthesis
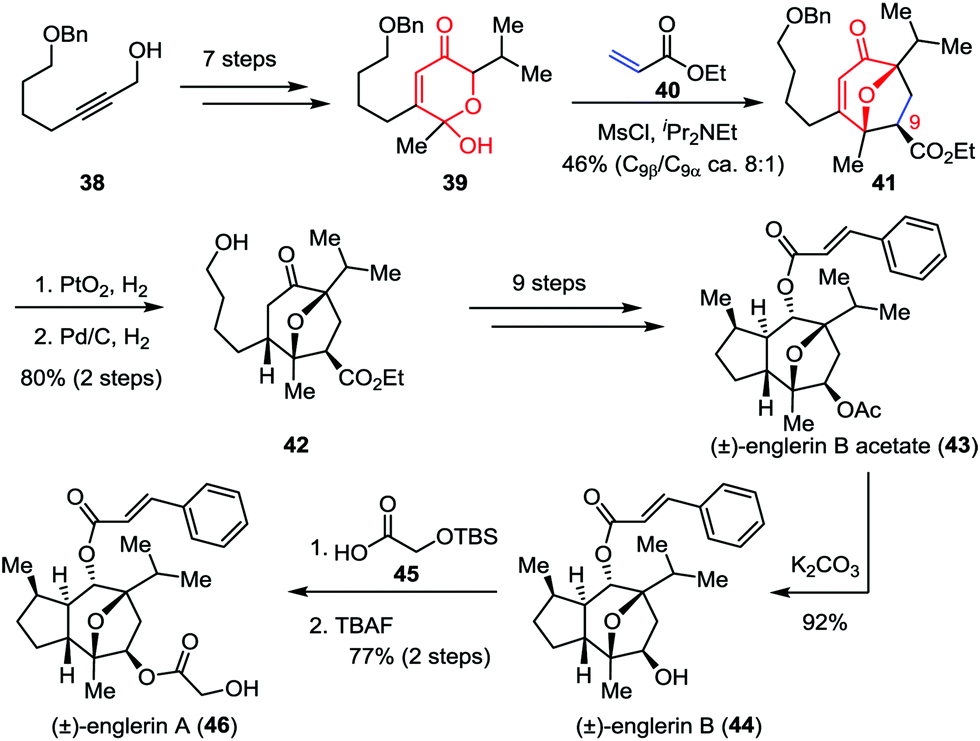
Isolated from the stem bark of Phyllanthus engleri, englerins A and B belong to the guaiane sesquiterpene family and exhibit potent and selective growth inhibitory activities against renal cancer cell lines.26 In 2011, Nicolaou, Chen and co-workers reported the racemic total synthesis of these two molecules.27 As shown in Scheme 8, lactol 39 was prepared from readily available starting material 38 within seven steps. The crucial [5+2] cycloaddition between 39 and ethyl acrylate 40 successfully delivered oxabicyclic enone 41 and its C9-epimer in 46% combined yield (8:1 dr). Subsequently, the sequential hydrogenative reduction of the olefin moiety and removal of the benzyl group afforded 42 in 80% yield, which was then transformed to englerin B acetate (43) through several conventional operations. Further elaboration of 43 led to englerins A (46) and B (44) with high efficiency.
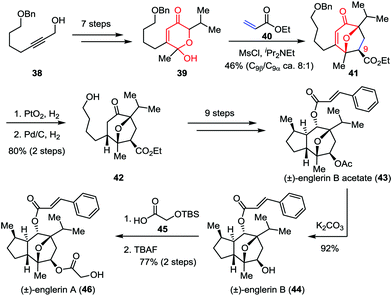 |
| | Scheme 8 Total synthesis of englerins A and B by Nicolaou, Chen and co-workers. | |
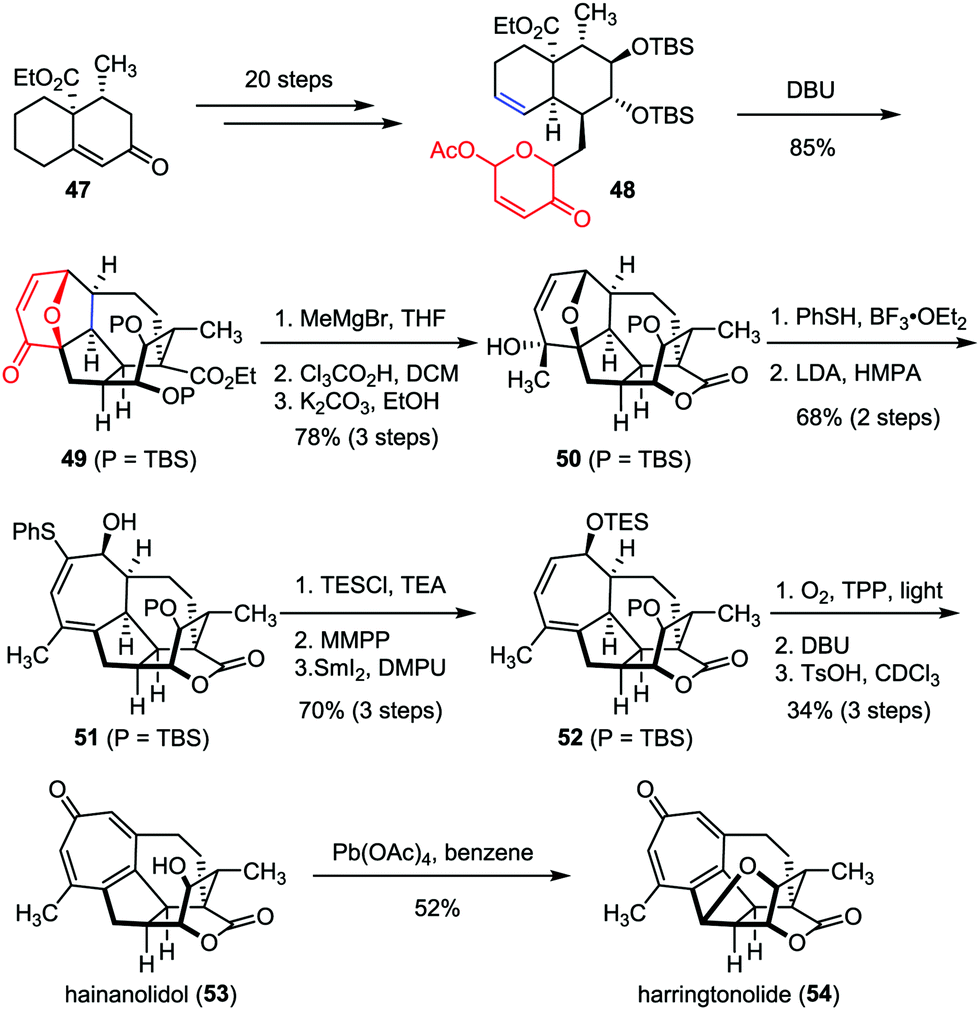
As two typical members of Cephalotaxus norditerpenes, harringtonolide and hainanolidol were isolated from C. harringtonia28 and C. hainanensis,29 respectively. Preliminary bioactivity investigations revealed that harringtonolide possesses antiviral and antineoplastic properties, while hainanolidol without the tetrahydrofuran ring was found inactive.30 In 2013, Tang and co-workers achieved the efficient construction of the core structure of harringtonolide and hainanolidol via an intramolecular oxidopyrylium-mediated [5+2] cycloaddition.31 As shown in Scheme 9, the anticipated cycloaddition of 48, which was prepared from enone 47 in twenty steps, proceeded smoothly in the presence of DBU, providing the adduct 49 as a single diastereomer in 85% yield. Stereoselective Grignard 1,2-addition of 49 followed by desilylation-induced lactonization afforded hexacycle 50. Subsequently, a two-step sequence was employed to cleave the ether bridge of 50 and gave diene 51 in 68% yield. Finally, hainanolidol (53) and harringtonolide (54) were obtained through a series of late-stage functionalizations involving a sequential [4+2] cycloaddition, a Kornblum–DeLaMare rearrangement and double elimination.
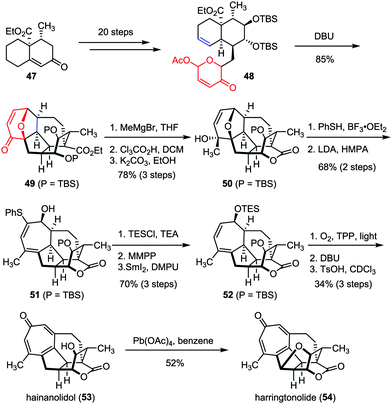 |
| | Scheme 9 Total synthesis of hainanolidol and harringtonolide by Tang and co-workers. | |
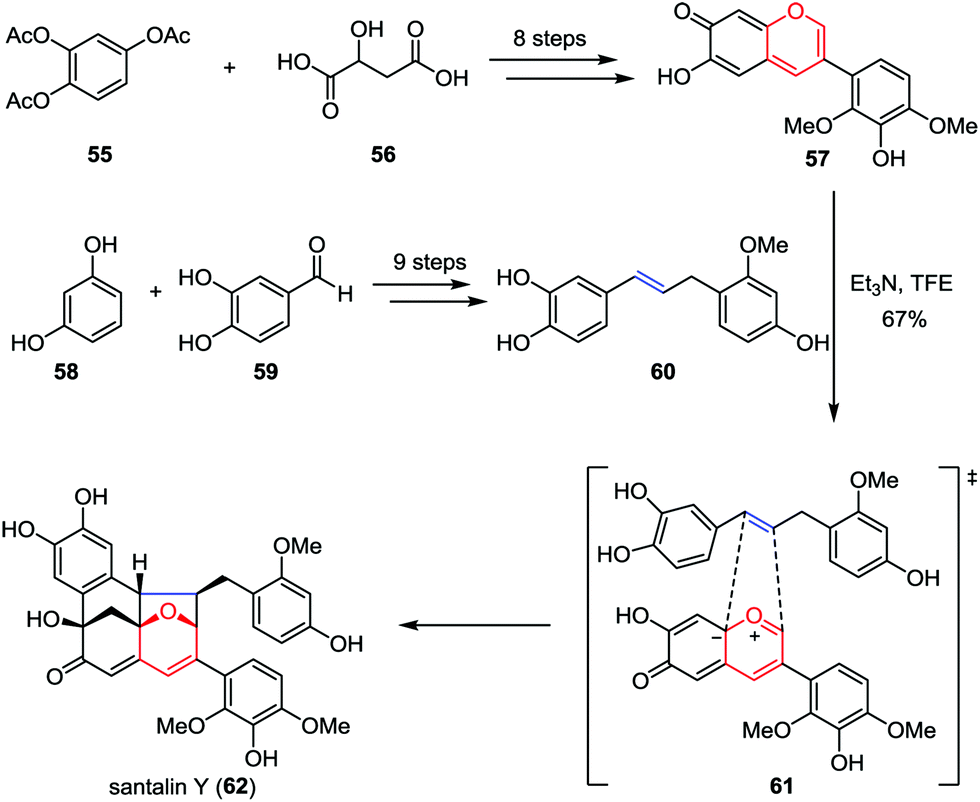
Santalin Y, a novel yellow pigment, was isolated from Pterocarpus santalinus by Nohara and co-workers in 1995.32 It possesses a unique [6,6,6,5]-oxafenestrane skeleton, which features a catechol moiety and partially methylated pyrogallol and resorcinol substituents. Recently, Trauner and Tantillo disclosed a biomimetic total synthesis of santalin Y (Scheme 10).33 Their synthesis commenced by aiming at the construction of anhydrobase 57, which was prepared from 1,2,4-triacetoxybenzene 55 and malic acid 56 in eight steps.34 On the other hand, the requisite benzylstyrene 60 could be obtained from readily available resorcinol 58 and protocatechualdehyde 59 by using olefin cross-metathesis as a key step. Upon treatment of a solution of 57 and 60 in trifluoroethanol (c = 0.06 M) at the solubility limits of these two intermediates with triethylamine at −20 °C to initiate the oxidopyrylium-mediated [5+2] cycloaddition, racemic santalin Y (62) was formed in 67% isolated yield.
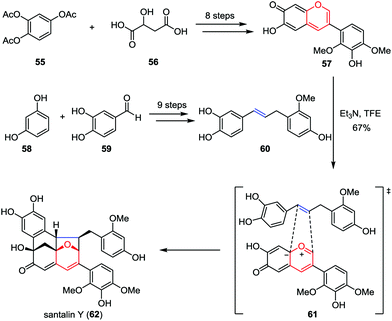 |
| | Scheme 10 Biomimetic total synthesis of santalin Y by Trauner and co-workers. | |
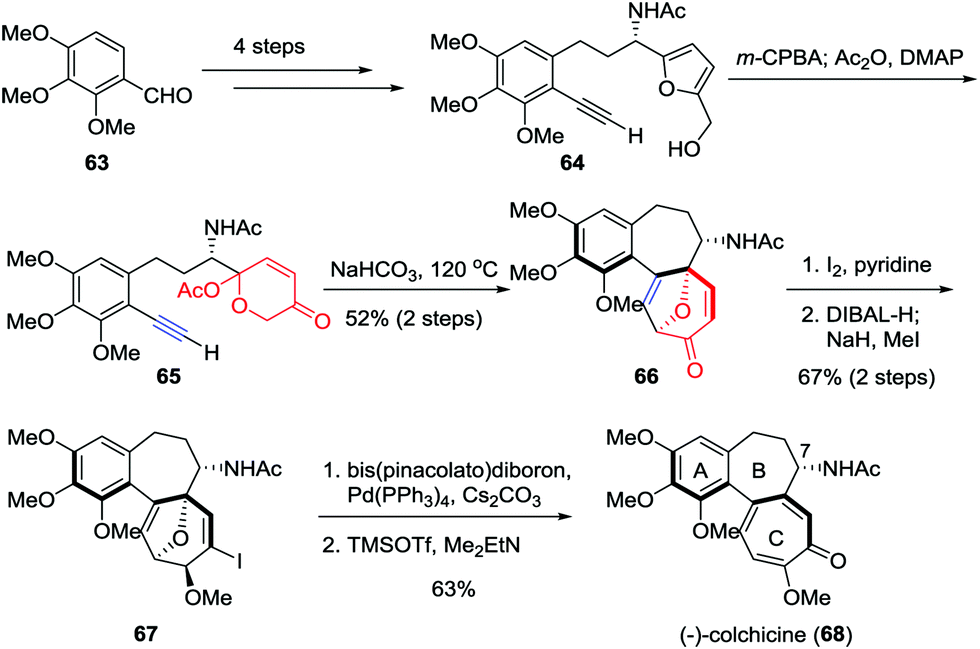
As the first tubulin-destabilizing agent reported in the literature, colchicine exhibits remarkable antimitotic activity that has been identified by a variety of indications. Due to its curative ability, this alkaloid is widely employed for the treatment of acute gout, familial Mediterranean fever and chronic myelocytic leukemia.35 Moreover, it has also been used as a neurotoxin in animal models in the research of Alzheimer's disease and epilepsy.36 Structurally, colchicine contains an unusual [6,7,7] tricyclic system. Furthermore, the single stereocenter at C7 as well as the aR-configured stereogenic axis defined by the pivot bond joining the A and C rings represents a formidable synthetic challenge. In 2017, Li and co-workers reported an enantioselective and scalable synthesis of (−)-colchicine (9.2% overall yield, >99% ee).37 As shown in Scheme 11, their total synthesis began with the preparation of furan-alkyne 64, which was realized in gram scale from aldehyde 63 over four steps. Subsequently, a two-step sequence involving Achmatowicz rearrangement and the oxidopyrylium-mediated [5+2] cycloaddition was utilized to build up the [6,7,7] tricyclic core structure of colchicinoids, providing 66 in 52% yield. The α-iodonization of 66 followed by ketone reduction/methylation resulted in the formation of 67 in 67% yield. Finally, the total synthesis of (−)-colchicine (68) was accomplished through a Suzuki–Miyaura cross coupling and double elimination of the oxa-bridge.
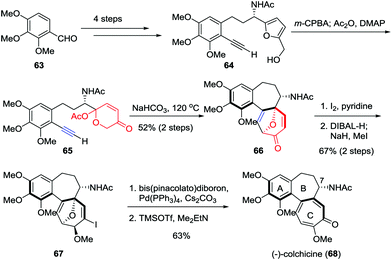 |
| | Scheme 11 Enantioselective synthesis of (−)-colchicine by Li and co-workers. | |
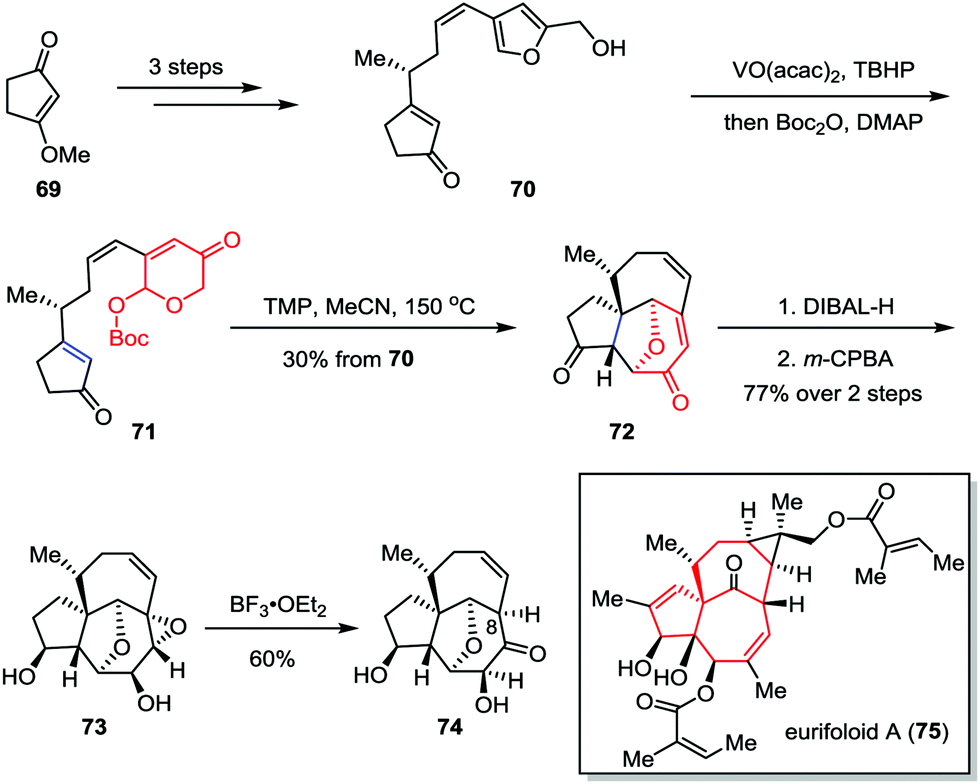
Eurifoloid A is a highly oxygenated tetracyclic diterpenoid, which was isolated from Euphorbia neriifolia in 2014.38 Structurally, eurifoloid A features a congested stereogenic cis-triol segment as well as an unusual and highly strained bicyclo[4.4.1]undecane ring system with trans-bridgehead (also known as in/out stereochemistry), which posed considerable synthetic challenges. In 2017, Li and co-workers described the synthetic studies toward eurifoloid A (Scheme 12).39 Their synthesis commenced with the preparation of furan 70, which was derived from 3-methoxycyclopent-2-en-1-one 69 in three steps. The Achmatowicz rearrangement of 70 using VO(acac)2 and TBHP followed by a one-pot Boc-protection of the resultant anomeric hydroxy group gave 71. The latter compound then underwent an intramolecular type II [5+2] cycloaddition to afford [5,7,7] tricycle 72 in 30% yield over two steps. Diastereoselective reduction of diketone followed by regio- and facial selective epoxidation delivered epoxide 73 as a single isomer in 77% yield over two steps. Finally, treatment of 73 with BF3·OEt2 afforded 74 in 60% yield. Although the Meinwald rearrangement of 73 gave the core structure of eurifoloid A (75) with undesired stereochemistry at C8, the above outcomes provided useful information for the design of new synthetic approach to this natural product.
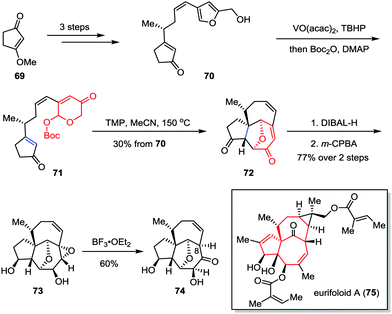 |
| | Scheme 12 Synthetic studies toward eurifoloid A by Li and co-workers. | |
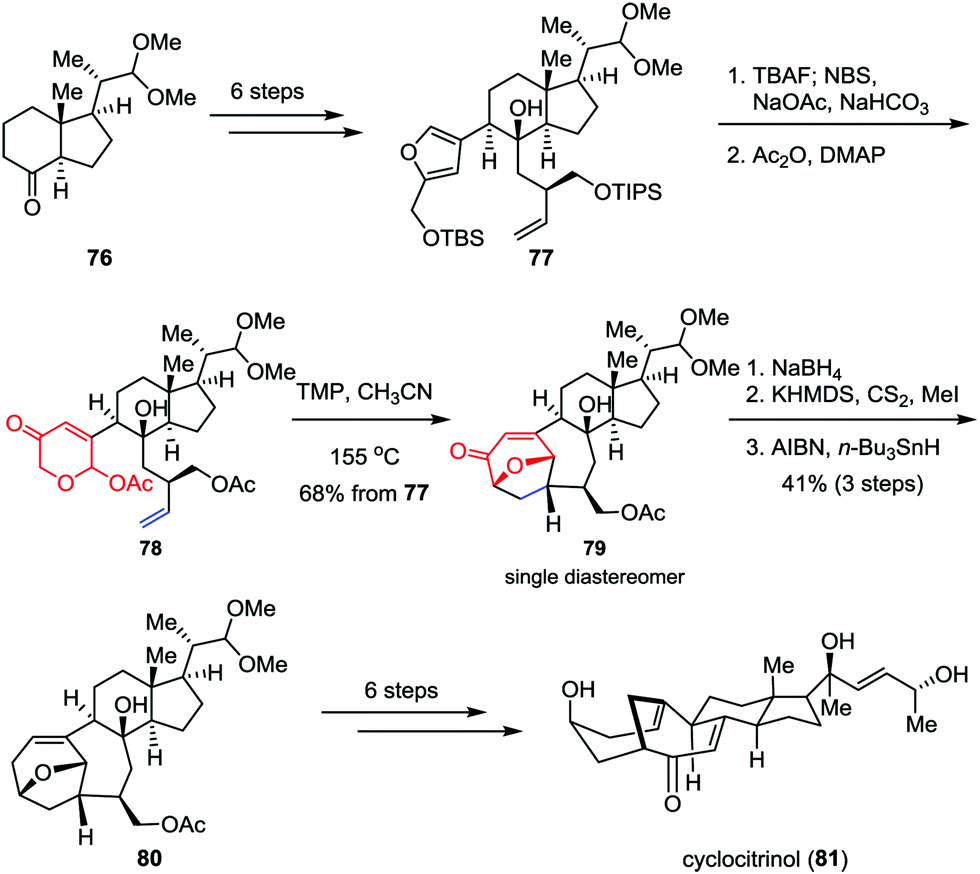
As an unusual C25 steroid, cyclocitrinol contains an unprecedented bicyclo[4.4.1]undecane ring system and a strained anti-Bredt bridgehead double bond. In preliminary studies, cyclocitrinol showed some interesting biological activities. For example, it can induce the generation of cAMP in GPR12-transfected CHO cells at 10 μM.40 However, the relative scarcity of this molecule from natural sources has impeded its further biological evaluation progress. Recently, Li and co-workers achieved the first and asymmetric total synthesis of cyclocitrinol (Scheme 13).41 Their synthesis began with the preparation of chiral furan 77, which could be derived from the commercially available starting material 76 in six steps. Desilylation of 77 followed by Achmatowicz rearrangement and acetylation generated the key precursor 78 smoothly. The intramolecular type II [5+2] cycloaddition was achieved by using TMP (2,2,6,6-tetramethylpiperidine) as a base in sealed tube and provided pentacycle 79 as a single diastereomer in 68% overall yield over three steps. The remaining ketone was then removed through 1,2-reduction followed by Barton deoxygenation, affording 80 in 41% yield over three steps. Finally, cyclocitrinol (81) was accessed through several conventional manipulations.
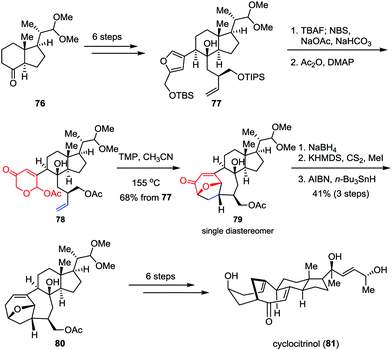 |
| | Scheme 13 Asymmetric total synthesis of cyclocitrinol by Li and co-workers. | |
3. [5+2] cycloaddition of pyridinium and quinolinium ylides
3.1 Rh-catalyzed [5+2] cycloaddition of pyridinium ylides
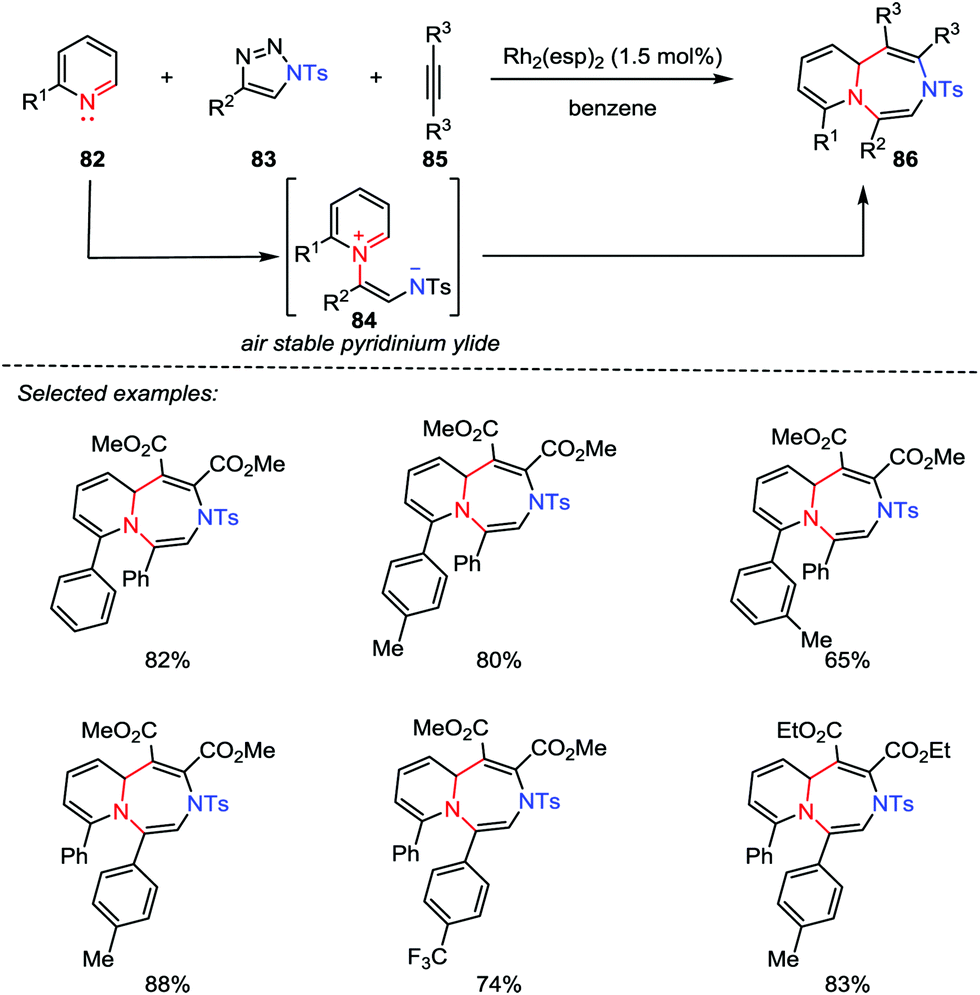
Although great progress has been made for the preparation of five- and six-membered heterocycles through catalytic [3+2] and [4+2] cycloadditions, the synthesis of seven-membered rings by using [5+2] cycloaddition reactions has not been investigated thus far ever since the seminal work of Wender et al. in 2002.42 Recently, Yoo and co-workers reported the first example of Rh-catalyzed multicomponent aza-[5+2] cycloaddition between the pyridines 82, 1-sulfonyl-1,2,3-trizoles 83 and alkynes 85. A variety of 1,4-diazepines 86 were obtained in good to excellent yields (Scheme 14).43 Mechanistically, with the use of a catalytic rhodium catalyst, a stable pyridinium ylide 84 was generated, which acted as a 1,5-dipole to react with the activated alkynes 85.
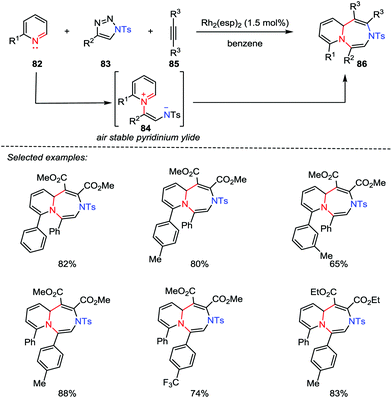 |
| | Scheme 14 The multicomponent aza-[5+2] cycloaddition by Yoo and co-workers. | |
3.2 Gold-catalyzed [5+2] cycloaddition of quinolinium ylide
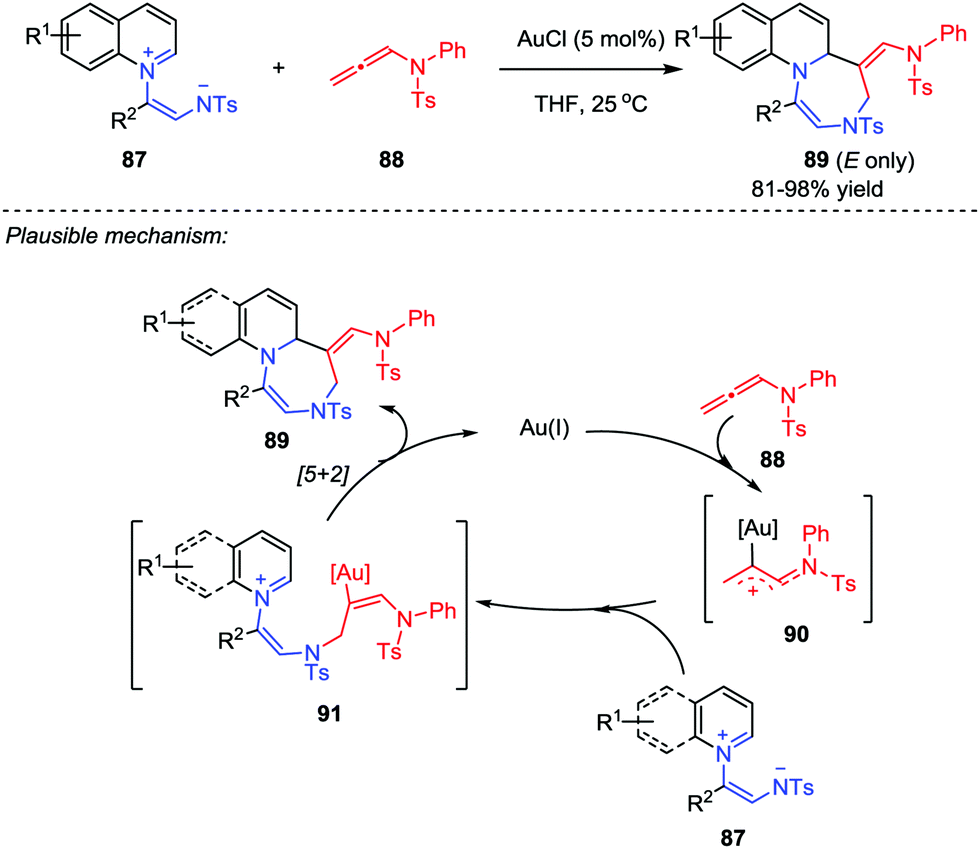
Based on their previous work on the [5+2] cycloaddition using a unique isolable pyridinium ylide as a 1,5-dipole, the Yoo group further developed a ligand-free gold-catalyzed [5+2] cycloaddition between quinolinium ylides 87 and allenamides 88 (Scheme 15).44 With the use of AuCl as catalyst, a series of stereospecific (E)-1,4-diazepines 89 were obtained in good to excellent yields. A plausible mechanism was proposed. Firstly, a gold-bound allylic cation 90 was generated by the activation of allenamide with gold catalyst, which was then attacked by the negative nitrogen of the quinolinium ylide 87 to yield the tethered intermediate 91. Subsequently, the intramolecular cyclization of 91 afforded the desired product and regenerated the catalyst. In addition, the described protocol could tolerate a broad scope of the substrates, whose potential and versatility has also been proved by a gram-scale feasibility test.
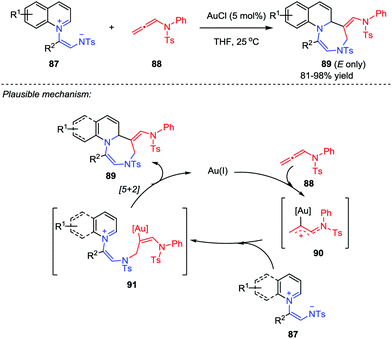 |
| | Scheme 15 The ligand free gold-catalyzed [5+2] cycloaddition by Yoo and co-workers. | |
3.3 Natural product synthesis
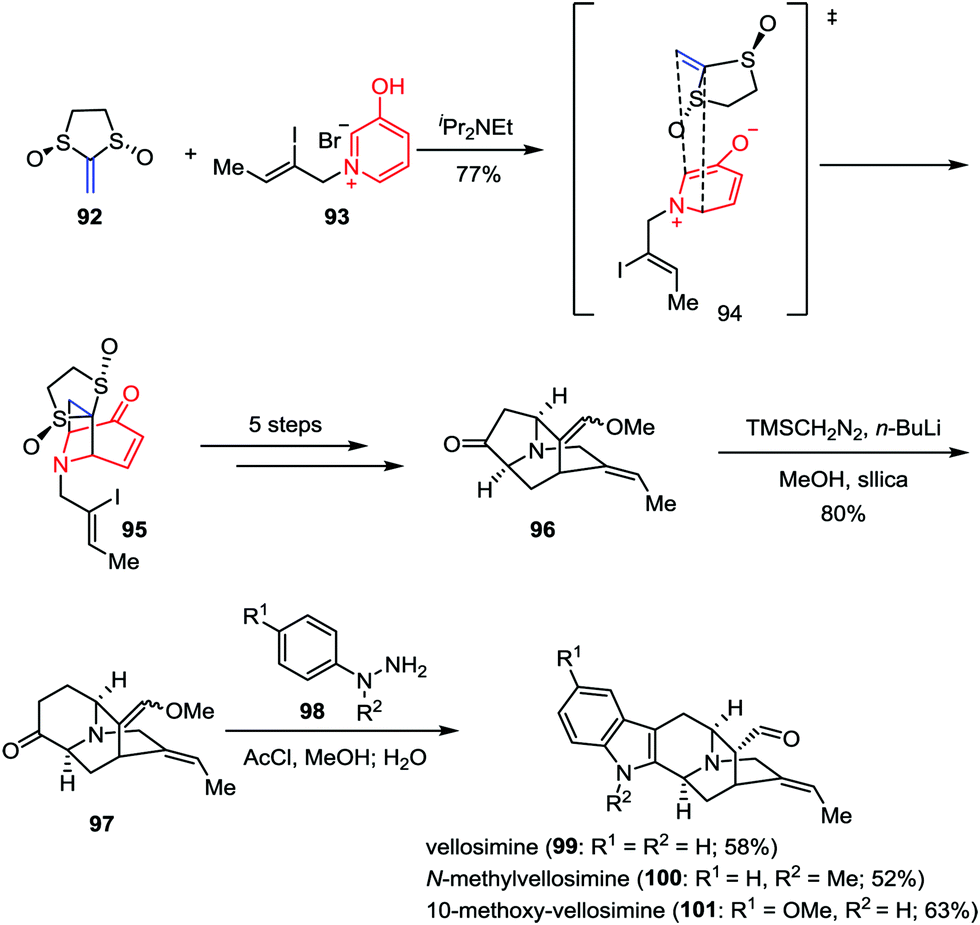
To date, more than ninety congeners of the sarpagine family have been isolated from the plant Apocynaceae.45 Due to the scarcity of these alkaloids and the lack of the synthetic analogues, the biological potential of these intricate architectures has rarely been investigated. In 2015, Gaich and co-workers demonstrated a general synthetic route to sarpagine alkaloids and completed the enantioselective total synthesis of vellosimine (99), N-methylvellosimine (100) and 10-methoxyvellosimine (101) in a concise manner (Scheme 16).46 Their synthesis commenced with a [5+2] cycloaddition between oxidopyridinium ion 93 and Aggarwal's chiral ketene equivalent 92 (93% ee),47 which provided the desired cycloadduct 95 and its regioisomer as a 2:1 mixture in 77% combined yield. After being converted to ketone 96 in five steps, the following ring enlargement proceeded smoothly and regioselectively, leading to the formation of 97 in 80% yield. Finally, vellosimine (99), N-methylvellosimine (100) and 10-methoxyvellosimine (101) were synthesized with high yields through Fischer indole synthesis by treatment of 97 with phenylhydrazines 98 bearing different substitution patterns.
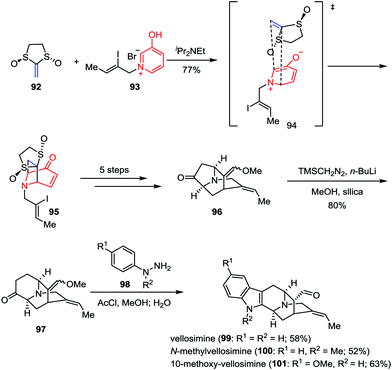 |
| | Scheme 16 Total synthesis of sarpagine alkaloids by Gaich and co-workers. | |
4. Perezone-type [5+2] cycloaddition
4.1 Oxidative dearomatization-induced [5+2] cascade reaction
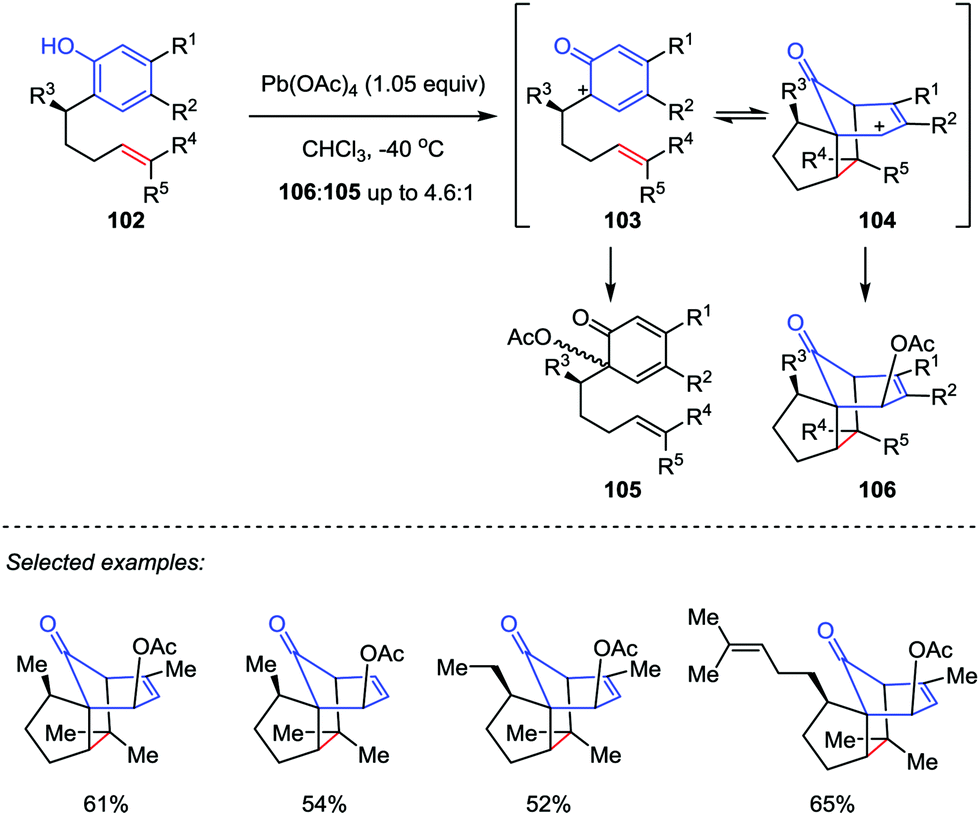
Inspired by the proposed biosynthesis of cedrol, Pettus and co-workers reported an efficient method for the construction of the tricyclo[5.3.1.01,5]undecane skeletons from curcuphenol derivatives (Scheme 17).48 The cascade reaction began with the oxidative dearomatization of ortho-(pent-4-enyl)-phenols 102 followed by an intramolecular [5+2] cycloaddition of the corresponding phenoxonium intermediate 103 with tethered olefin by using Pb(OAc)4 as an oxidant. The benzylic substituents and trisubstituted olefin subunits were found critical to the success of the oxidative dearomatization-induced (ODI) [5+2] cascade reaction, which afforded the cedrene-type skeletons 106 in good to moderate yields with high diastereoselectivities. Otherwise, the reaction gave the ortho-acetoxylated compounds 105 as the major product.
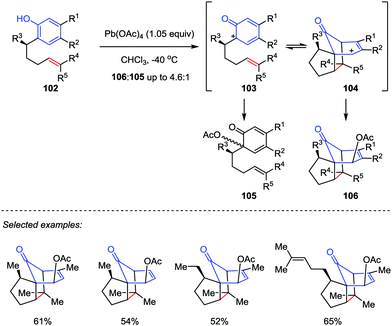 |
| | Scheme 17 ODI-[5+2] cycloaddition by Pettus and co-workers. | |
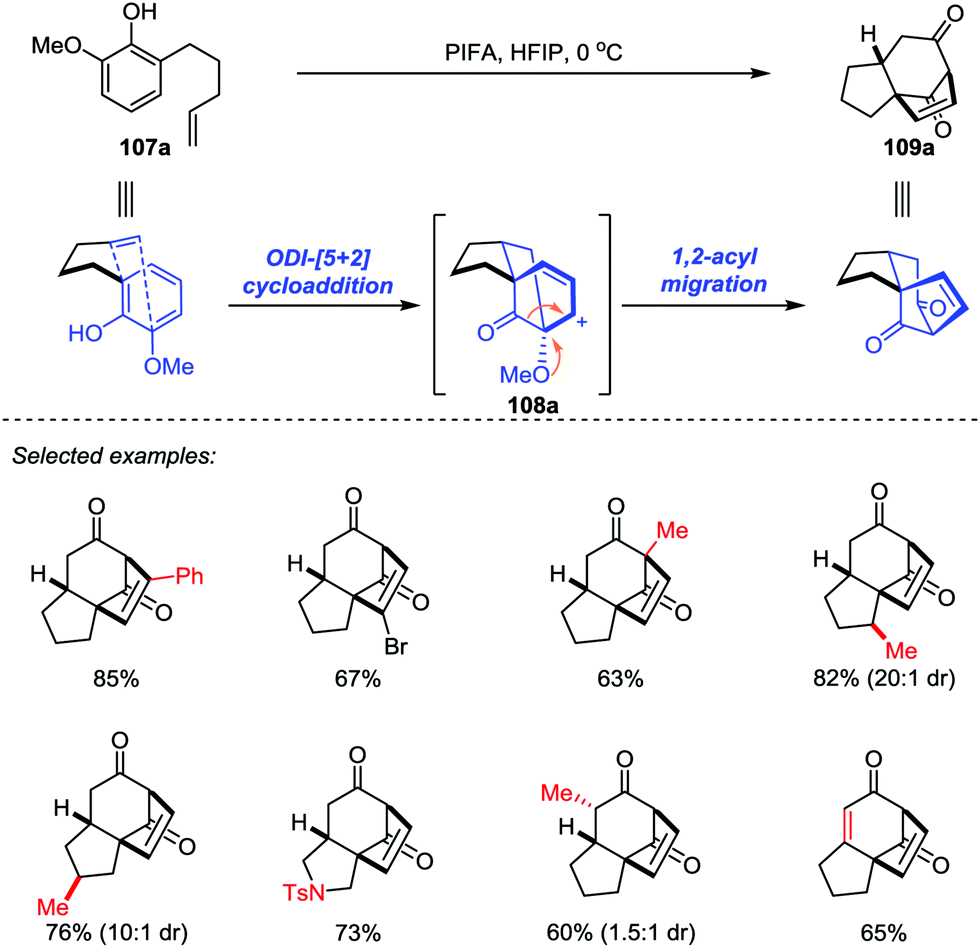
In 2017, Ding and co-workers developed an ODI-[5+2] cycloaddition/pinacol-type 1,2-acyl migration cascade reaction to assemble the highly oxygenated bicyclo[3.2.1]octane system (Scheme 18).49 With the use of PhI(CF3CO2)2 (PIFA) as the oxidant and 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as the solvent, various vinylphenols 107 with different substituents on the aromatic ring or functionalities on the side chain such as monosubstituted and disubstituted alkenes, and even alkynes were transformed into the respective products 109 with up to 85% yield. Mechanistically, the cationic cedrene-type skeleton intermediate was generated through an ODI-[5+2] cycloaddition and the preinstalled methoxy group at the ortho position of the phenol promoted the pinacol-type 1,2-acyl migration to forge the desired ent-kaurene-type skeleton.
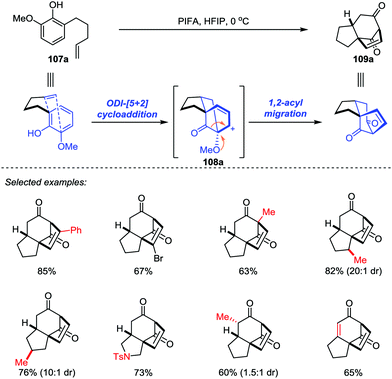 |
| | Scheme 18 ODI-[5+2] cycloaddition/pinacol-type 1,2-acyl migration cascade by Ding and co-workers. | |
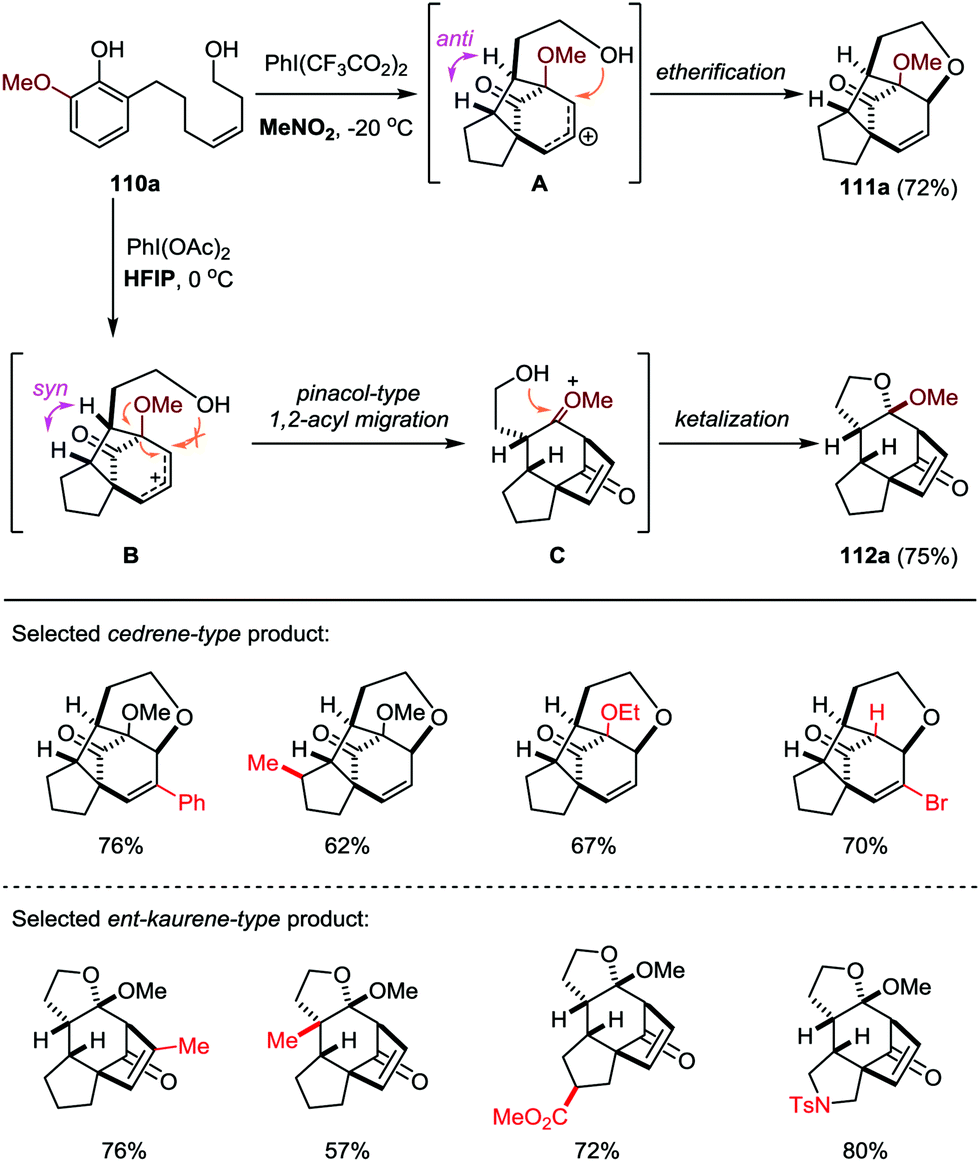
Based on this work, the Ding group further developed a solvent-dependent ODI-[5+2] cascade reaction in the presence of hypervalent iodine oxidants (such as PhI(OAc)2 and PhI(CF3CO2)2), which led to the facile and divergent construction of functionalized cedrene and ent-kaurene-type skeletons with good yields and excellent diastereoselectivities (Scheme 19).50 By employment of MeNO2 as the solvent, the cedrene-type product 111a was obtained through an ODI-[5+2] cycloaddition followed by interception of the in situ generated allylic carbocation with the appendant hydroxyl group through an intramolecular etherification. On the other hand, with the use of HFIP as the solvent, the ent-kaurene-type product 112a was achieved through an ODI-[5+2] cycloaddition followed by pinacol-type 1,2-acyl migration and intramolecular ketalization.
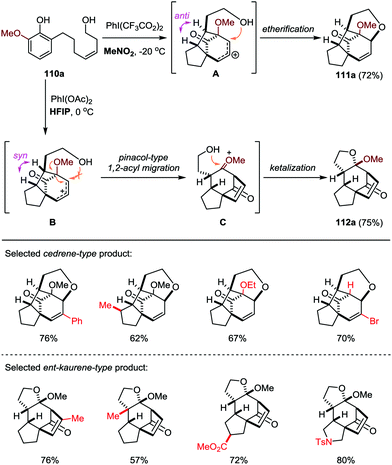 |
| | Scheme 19 ODI-[5+2] cascade approach for the divergent synthesis of cedrene and ent-kaurene-type product by Ding and co-workers. | |
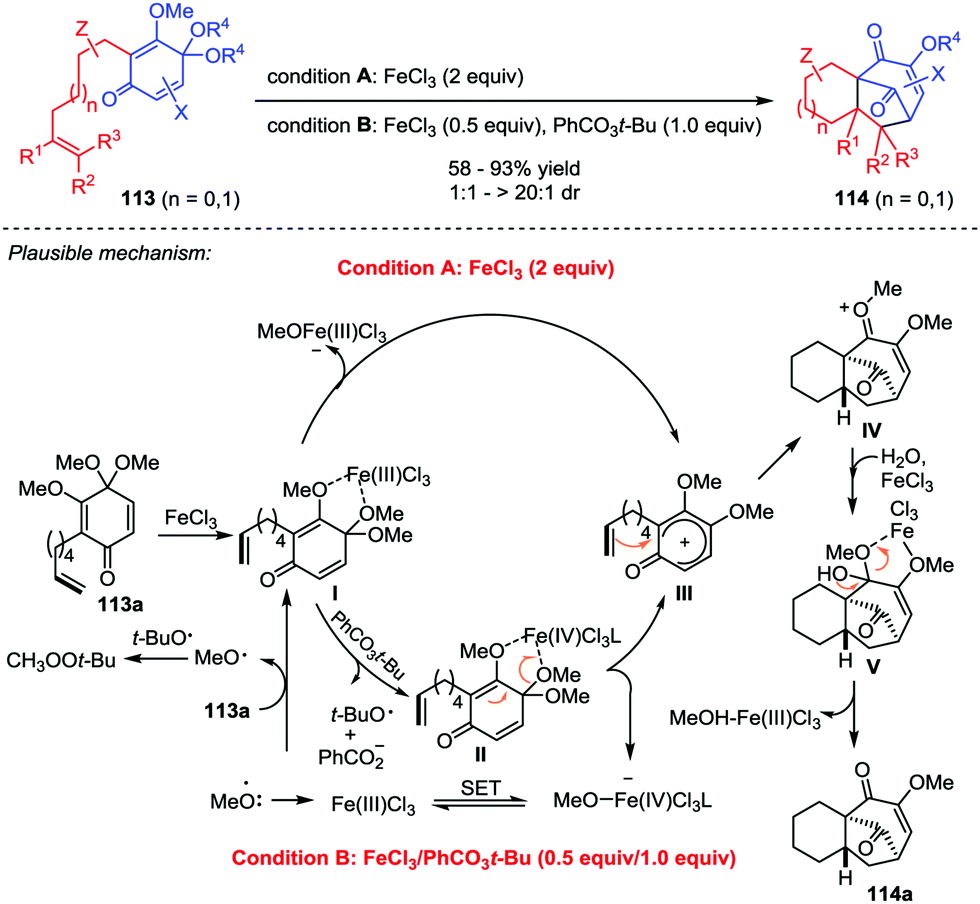
4.2 Iron-catalyzed [5+2] cycloaddition
Natural products and pharmaceutical intermediates containing diverse tricyclo[6.3.1.01,6]dodecane core structures often exhibit remarkable biological activities.51 Recently, Liu and co-workers presented an iron-catalyzed perezone-type [5+2] cycloaddition reaction of substrate 113 to afford tricyclo[6.3.1.01,6]dodecane scaffolds 114, which created two new C–C bonds and four stereogenic centers in one single operation with good yields and excellent dr values (Scheme 20).52 The products were accessible with two different reaction conditions, stoichiometric FeCl3 (2 equiv.) and catalytic FeCl3/PhCO3t-Bu (0.5/1 equiv.). Mechanistically, the reaction of 113a for both conditions might share the same pathway involving the iron-mediated generation of a carbocation intermediate III, which triggered the subsequent [5+2] cycloaddition to give intermediate IV. Finally, demethoxylation of IV with the aid of H2O and FeCl3 furnished 114a.
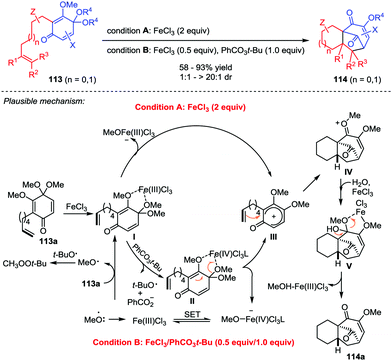 |
| | Scheme 20 [5+2] Cycloaddition toward tricyclo[6.3.1.01,6] skeleton by Liu and co-workers. | |
4.3 Natural product synthesis
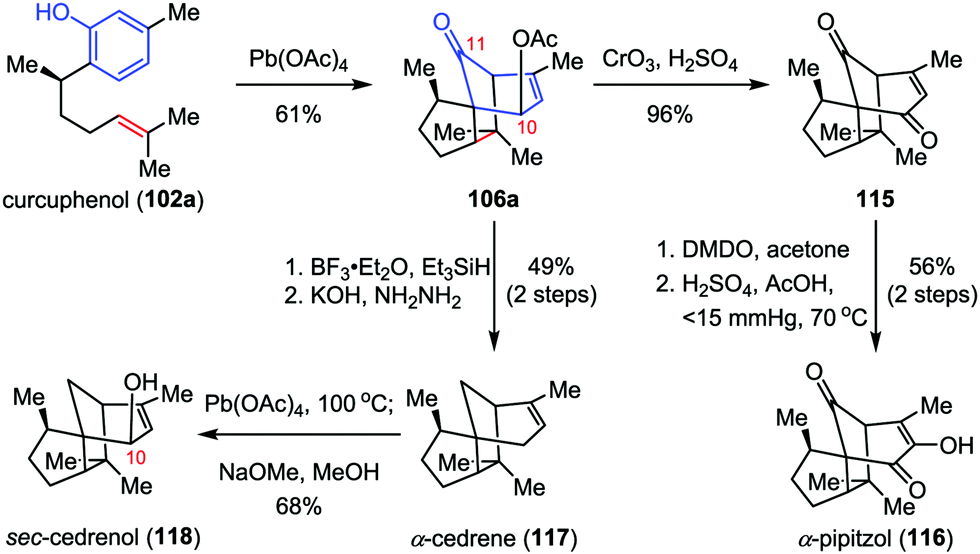
α-Cedrene, α-pipitzol and sec-cedrenol are important members of the cendranoid family, which possess a characteristic tricyclo[5.3.1.01,5]undecane skeleton. α-Cedrene (117) was isolated from the American red cedar Juniperus virginiana,53 which shows antimicrobial, insecticidal and termiticidal activities54 and has been used as an approved food preservative.55 α-Pipitzol (116) was isolated from the root extracts of Perezias cuernavacana.56sec-Cedrenol (118), a relative newcomer to the cedranoid family, was produced by the Rhodococcus bacterium sp. KSM-7358 upon exposure to feed stocks of α-cedrene57 and displays a potent stimulant of the histamine H3 receptor, which might be useful for the cure of bronchial asthma, hyperlipidemia, and inflammation.58 In 2010, Pettus and co-workers described the total synthesis of these natural products from curcuphenol 102a, featuring an ODI-[5+2] cascade in the construction of the common intermediate 106a (Scheme 21).48 Jones oxidation of 106a afforded enone 115 in 96% yield. Subsequently, epoxidation of 115 with DMDO followed by ring-opening of the α,β-epoxyketones intermediate with Hu's mixed-acid catalyst (H2SO4, AcOH, 70 °C, reduced pressure) provided the desired α-pipitzol (116) in 56% yield over two steps. Erasure of the acetate functionality at C10 of 106a by Et3SiH/BF3·OEt2 and Wolff–Kishner reduction delivered α-cedrene (117) in 49% yield over two steps. Finally, further allylic oxidation of 117 at C10 by Pd(OAc)4 furnished sec-cedrenol (118) in 68% yield.
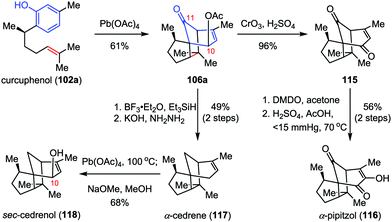 |
| | Scheme 21 Total synthesis of α-cedrene, α-pipitzol and sec-cedrenol by Pettus and co-workers. | |
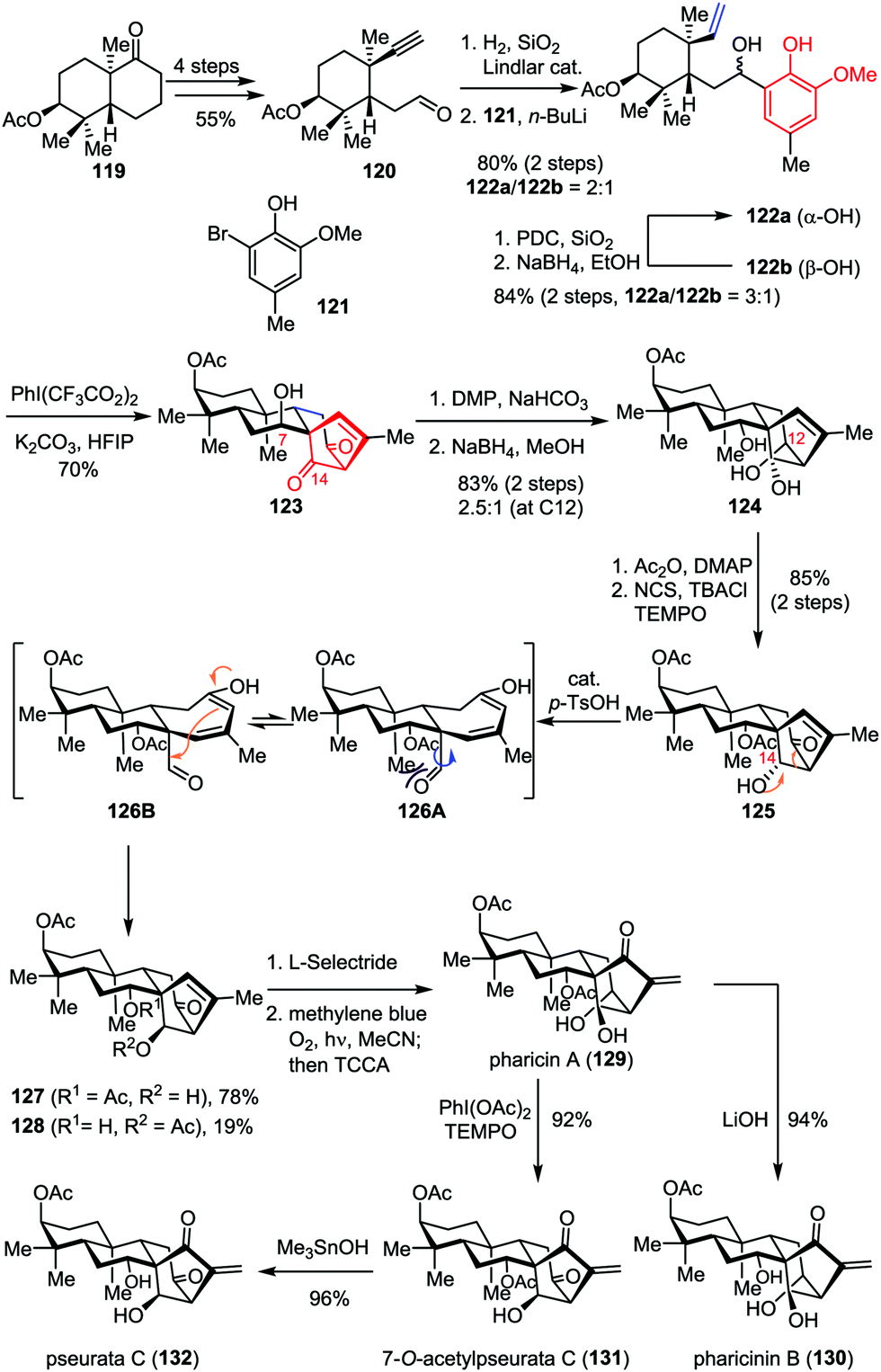
ent-Kaurene diterpenoids display a wide range of bio-activities such as antimalarial, antitumor and antiviral properties.59 Among them, pharicin A (129) was found to induce tumor cell apoptosis and suppress tumor growth.60 The construction of the bicyclo[3.2.1]octane carbon framework embedding in these molecules usually requires multiple reaction sequences, thus inevitably increased the functional group transformation and resulted in lengthier linear routes. Recently, Ding and co-workers reported the concise total synthesis of pharicin A and its congeners in a straightforward manner by using the ODI-[5+2] cycloaddition cascade as a key step (Scheme 22).49 Their synthesis commenced with the preparation of acetylene aldehyde 120, which was derived from the readily available bicyclic ketone 119 in four steps. Partial hydrogenation of the acetylene moiety with Lindlar catalyst followed by 1,2-addition with the organolithium reagent generated from bromophenol 121 and n-BuLi afforded a 2:1 mixture of vinylphenol 122a and 122b in 80% combined yield. The latter isomer could be recycled via an efficient oxidation/reduction sequence, affording a 3:1 mixture of diastereomers favoring 122a. Treatment of 122a with PhI(CF3CO2)2 and K2CO3 in HFIP delivered the desired tetracyclic diketone 123 as a single diastereomer in 70% yield even on gram scale. Subsequently, epimerization at C7 through Dess–Martin oxidation followed by global reduction provided triol 124 in 83% yield as a mixture of diastereomers (2.5:1 dr at C12). Unfortunately, the exclusive generation of the undesired α-OH at C14 was also observed. To address this problem, a two-step sequence involving monoacetylation at C7 and regioselective oxidation at C12 was firstly carried out, which delivered alcohol 125 in 85% yield. After an extensive investigation, it was found that the inversion of the stereochemistry at C14 proceeded smoothly in the presence of catalytic p-TsOH, delivering 127 in 78% yield through a retroaldo/aldol process. Finally, pharicin A (129) was achieved through a stereoselective ketone reduction and Schenck ene reaction. Further one or two-step elaboration of 129 led to the completion of the total synthesis of pharicinin B (130), 7-O-acetylpseurata C (131) and pseurata C (132) in high yields.
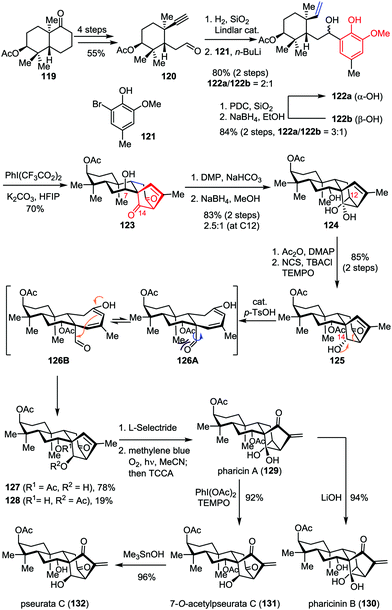 |
| | Scheme 22 Total synthesis of pharicin A, pharicin B, 7-O-acetylpseurata C and pseurata C by Ding and co-workers. | |
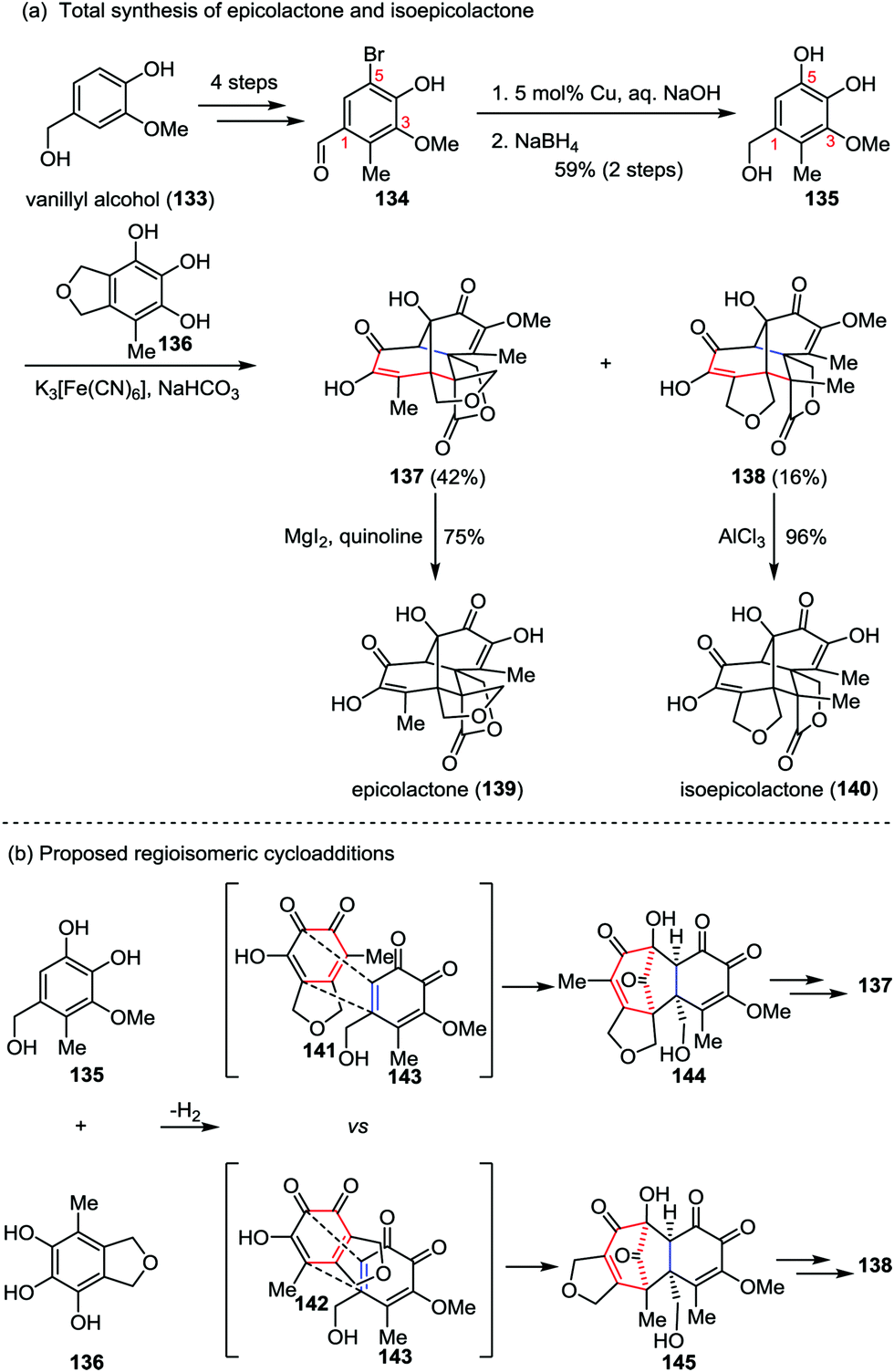
Epicolactone, a highly compact natural product, was isolated from endophytic fungi of the species Epicoccum.61 It exhibits both antimicrobial and antifungal activities. Trauner and co-workers described a biomimetic synthesis of epicolactone (139) in only eight steps from readily available vanillyl alcohol 133 (Scheme 23a).62 Their synthesis commenced with the preparation of bromobenzaldehyde 134, which was derived from vanillyl alcohol 133 in four steps through a reported procedure.63 Installation of another phenol hydroxyl group at 5-position of 134via copper-catalyzed hydroxylation followed by reduction of the aldehyde furnished catechol 135 in 59% yields over two steps. It should be noted that the existing of a phenolic methyl ether at 3-position of substrate 135 is crucial to the success of this approach, which reduced the possibility of dimerization and provided a stable intermediate in the next key step. The other partner, epicoccine 136, could be prepared in five steps from eudesmic acid on a multigram scale.63 Subjection of 135 to a buffer solution of K3[Fe(CN)6] followed by slow addition of 136 afforded epicolactone methyl ether 137 in 42% yield, along with the regioisomeric heterodimer 138 in 16% yield. Further demethylation of both isomers led to epicolactone (139) and isoepicolactone (140) in 75% and 96% yields, respectively.
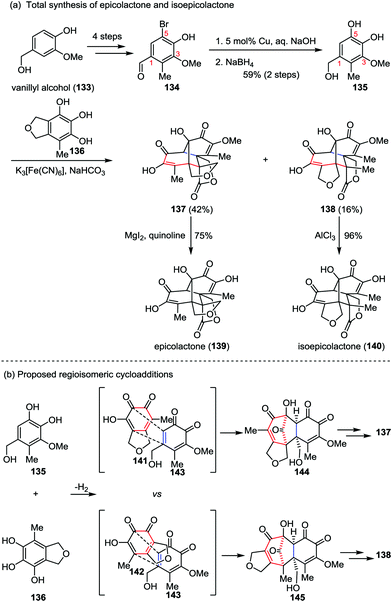 |
| | Scheme 23 (a) Total synthesis of epicolactone and isoepicolactone by Trauner and co-workers. (b) Proposed mechanism for the divergent [5+2] cycloaddition. | |
The features of the divergent [5+2] cycloaddition lie in two aspects: (1) the reaction proceeded under biomimetic conditions and finished in seconds; and (2) the complex caged product comprising five condensed rings bearing three contiguous quaternary carbons and a tertiary alcohol could be formed in a single operation from two relatively simple phenols. The initial [5+2] cycloaddition closely matches the proposed biosynthetic route while utilizing the methoxy o-quinone (143) instead of the unmasked hydroxy o-quinone (Scheme 23b).64 The cycloaddition led to the formation of carbonyl-bridged intermediate 144, and hence the following steps were all intramolecular, avoiding enormous steric hindrances. Similarly, the [5+2] cycloaddition of 143 with 142, a tautomer of 141, afforded an intermediate (145) en route to the minor regioisomer 138.
5. [5+2] Cycloaddition of vinyl/allenyl substituted three-membered rings
5.1 [5+2] cycloaddition of vinyl/allenyl cyclopropanes
Highly strained cyclopropane derivatives have served as useful and powerful C3 building blocks for the construction of various ring systems.65 In this area, the metal-catalyzed cleavage of the activated carbon–carbon σ bond of the cyclopropane ring is one of the most attractive methods from a synthetic point of view.66 Among them, the transition metal-catalyzed [5+2] cycloaddition of vinyl cyclopropanes (VCPs) is a straightforward way to access seven-membered carbocycles. In 1995, Wender and co-workers reported the first example of transition metal-catalyzed [5+2] cycloaddition between VCPs and alkynes, providing a conceptually new method for the synthesis of seven-membered rings.67 Since then, a variety of reactions concerning metal-catalyzed [5+2] cycloaddition of VCPs with different π-systems were successively reported.
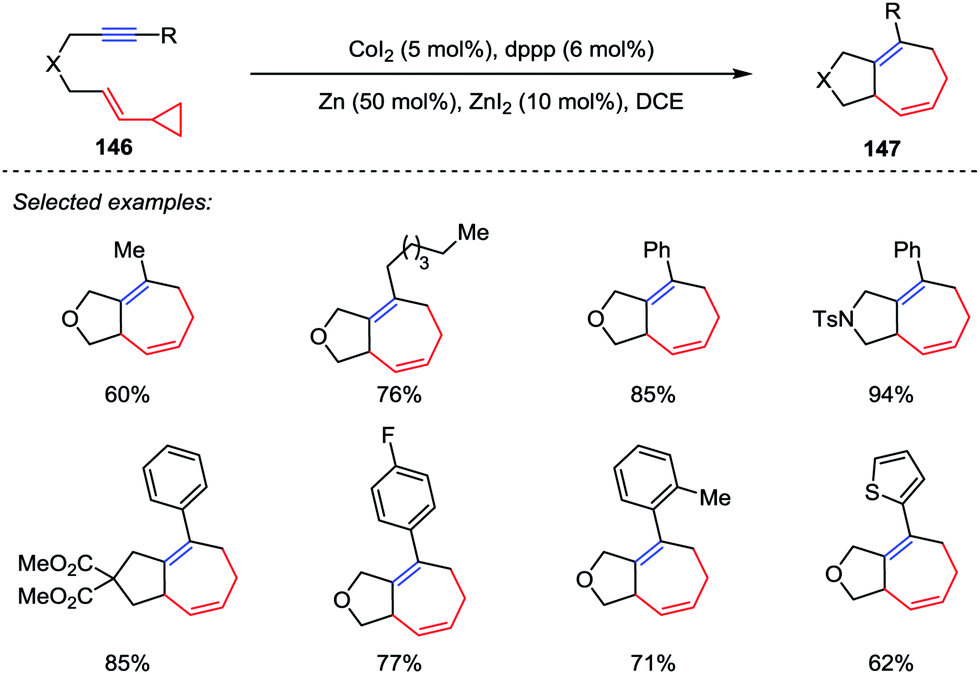
Among them, other transition-metals have also been successfully developed as catalysts in a number of intramolecular [5+2] cycloadditions recently, including ruthenium,68 nickel,69a iron,69b iridium.69c Recently, Yoshikai and co-workers addressed a cobalt-catalyzed intramolecular [5+2] cycloaddition reaction of VCPs and alkynes (Scheme 24).70 A wide range of tethered VCP/alkyne substrates 146 were tolerated by using the optimized catalytic conditions comprising CoI2 (5 mol%), dppp (6 mol%) and Zn dust as a reductant, thus providing the corresponding cycloheptadienes 147 in good to excellent yields.
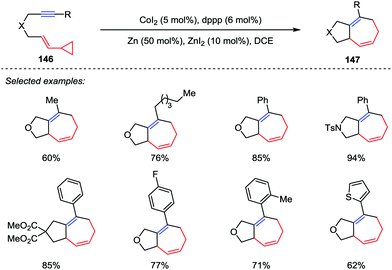 |
| | Scheme 24 Cobalt-catalyzed intramolecular [5+2] cycloadditions by Yoshikai and co-workers. | |
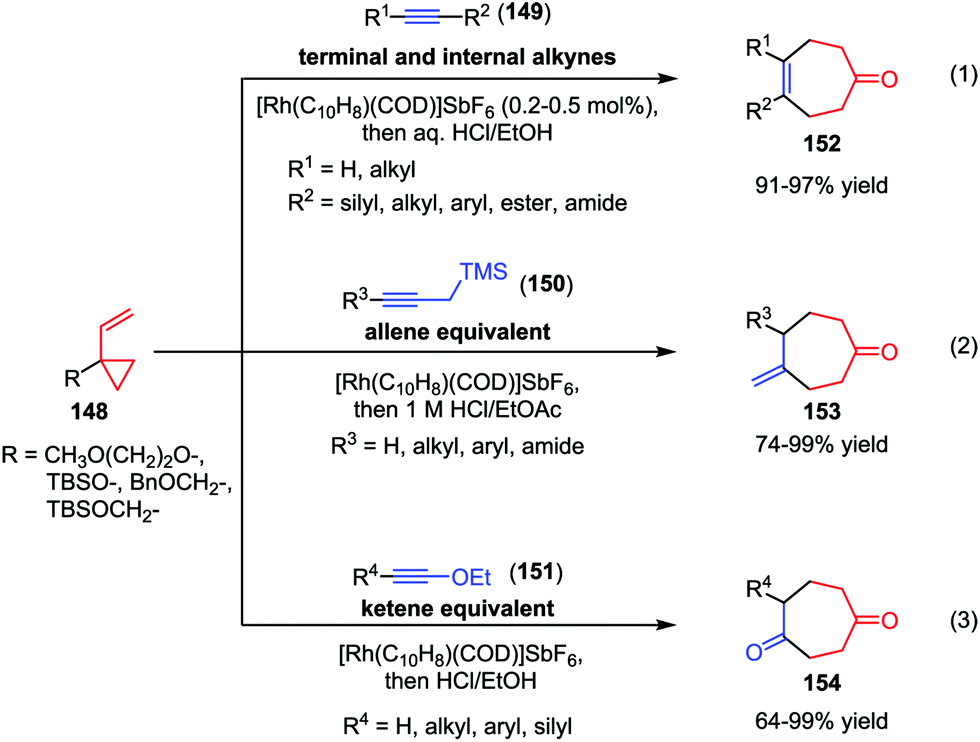
Recently, the Wender group made much progress in the area of intermolecular [5+2] cycloaddition of VCPs. In 2010, they described a cationic rhodium(I) complex-catalyzed intermolecular [5+2] cycloaddition of 1-alkoxy- or 1-alkyl-VCPs 148 with a wide range of terminal and internal alkynes 149 (Scheme 25).71a High efficiencies (91–97% yields) were observed under low catalyst loadings (0.2–0.5 mol%) and practical reaction concentrations (0.5–0.1 M) (eqn (1)). As conventional allenes are ineffective 2-carbon components in many intermolecular cycloadditions including metal-catalyzed [5+2] cycloadditions, the Wender group employed propargyl-trimethylsilanes (150) as allene surrogates and developed a stepwise intermolecular [5+2] cycloaddition/protodesilylation reactions, affording the corresponding cycloheptanone 153 in good to high yields (eqn (2)).71b In this work, 150 functioned as the safe, easily handled synthetic equivalents of gaseous allenes or monosubstituted allenes that are difficult to access. Furthermore, they also used ynol ethers 151 as ketene equivalents in the Rh-catalyzed intermolecular [5+2] cycloadditions with VCPs 148 (eqn (3)).71c These cycloaddition reactions proceeded smoothly at ambient temperature and afforded the substituted cyclohepta-1,4-diones 154 with up to 99% yield. In addition, a wide range of functionalities, such as chloro, methoxyl, and ester groups were tolerated in this reaction.
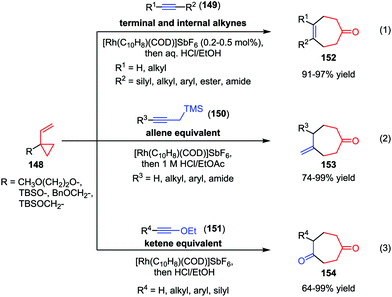 |
| | Scheme 25 [5+2] Cycloadditions of aklynes and VCPs by Wender and co-workers. | |
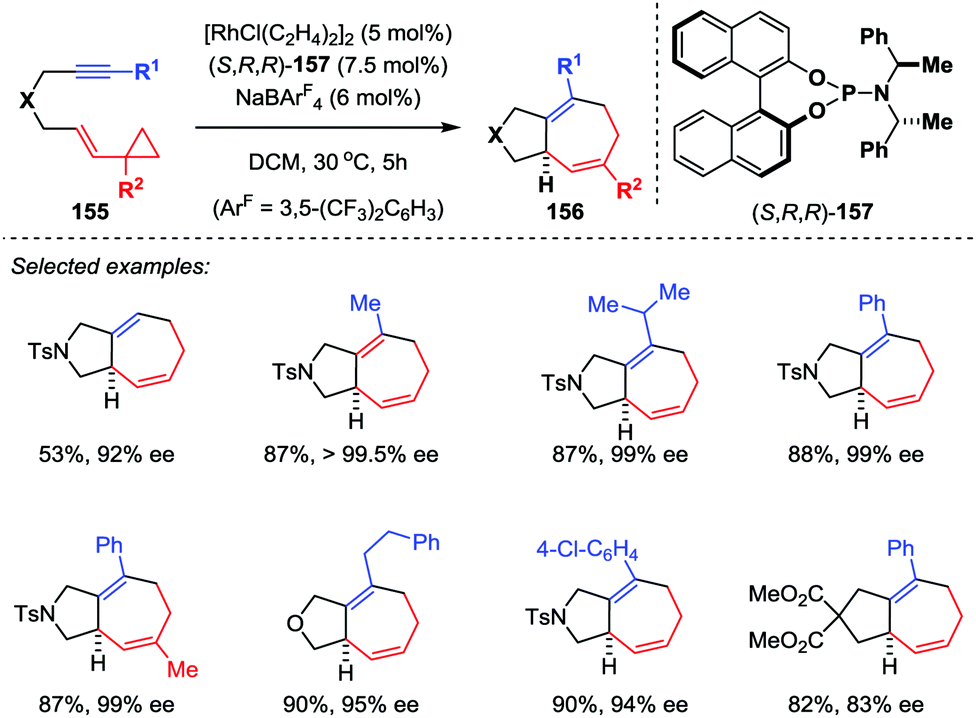
For asymmetric transition-metal-catalyzed [5+2] cycloadditions of VCPs, the catalyst-controlled method is envisioned more efficient and powerful than the substrate-controlled pathway. Wender and co-workers firstly used a rhodium-BINAP chiral catalyst in the asymmetric [5+2] cycloaddition reaction of VCPs and alkenes.72 In their results, asymmetric cycloaddition with alkenes could afford enantiomeric excesses as high as 99%. However, similar reactions with alkyne substrates were not as successful, giving only moderate enantioselectivities. To address this limitation, Shintani and Hayashi developed a highly efficient asymmetric intramolecular [5+2] cycloaddition of alkyne-VCPs 155 through rhodium catalysis (Scheme 26).73 After an extensive investigation, they found the combination of [RhCl(C2H4)2]2 with chiral phosphoramidite ligand 157 in the presence of NaBArF4 is the optimal condition, providing cycloadducts 156 with excellent enantioselectivities (up to 99.5% ee).
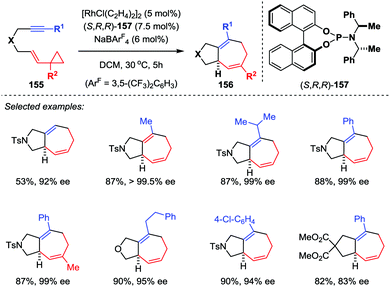 |
| | Scheme 26 Rh-catalyzed asymmetric [5+2] cycloadditions of alkyne-VCPs by Shintani and Hayashi. | |
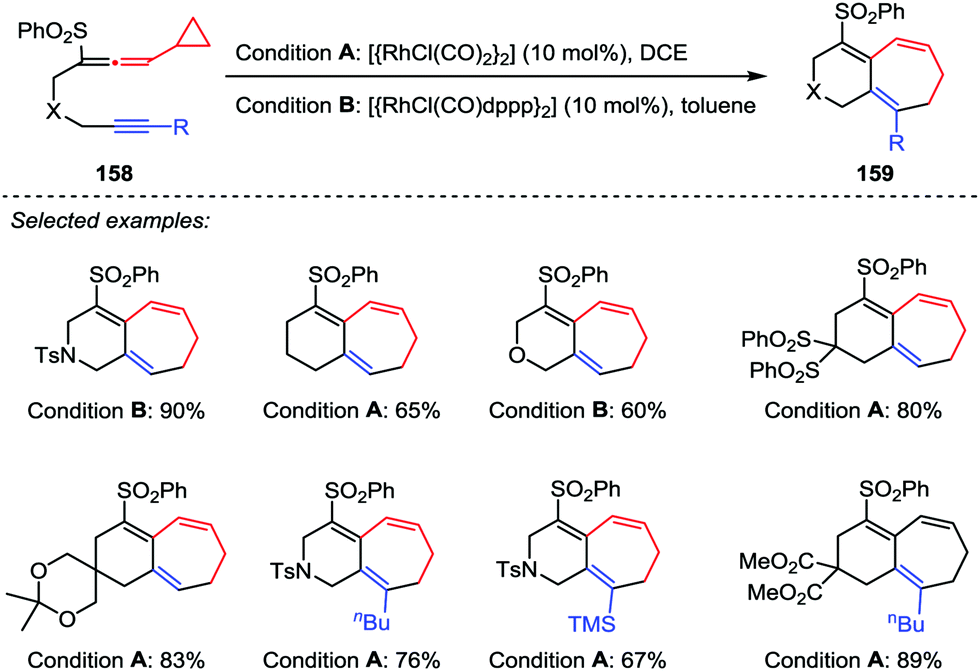
In 2010, Mukai et al. developed a Rh(I)-catalyzed intramolecular [5+2] cycloaddition reaction between phenylsulfonylallenes 158 containing a cyclopropyl group at the allene terminus and alkynes under mild conditions (Scheme 27).74 This reaction provided the desired products 159 in moderate to good yields with the reactants possessing an internal or terminal alkyne in the presence of [{RhCl(CO)2}2] or [{RhCl(CO)dppp}2]. The results are in sharp contrast with those results obtained from the [{RhCl(CO)2}2]-catalyzed [5+2] cycloaddition of alkyne-VCPs,75 in which an internal alkyne consistently and efficiently afforded the corresponding ring-closed product, whereas no [5+2] cycloaddition product was observed when a terminal alkyne was involved.
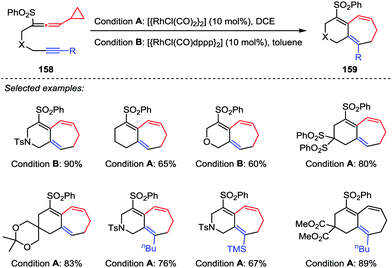 |
| | Scheme 27 [5+2] Cycloadditions of alkynes and allenyl cyclopropanes by Mukai and co-workers. | |
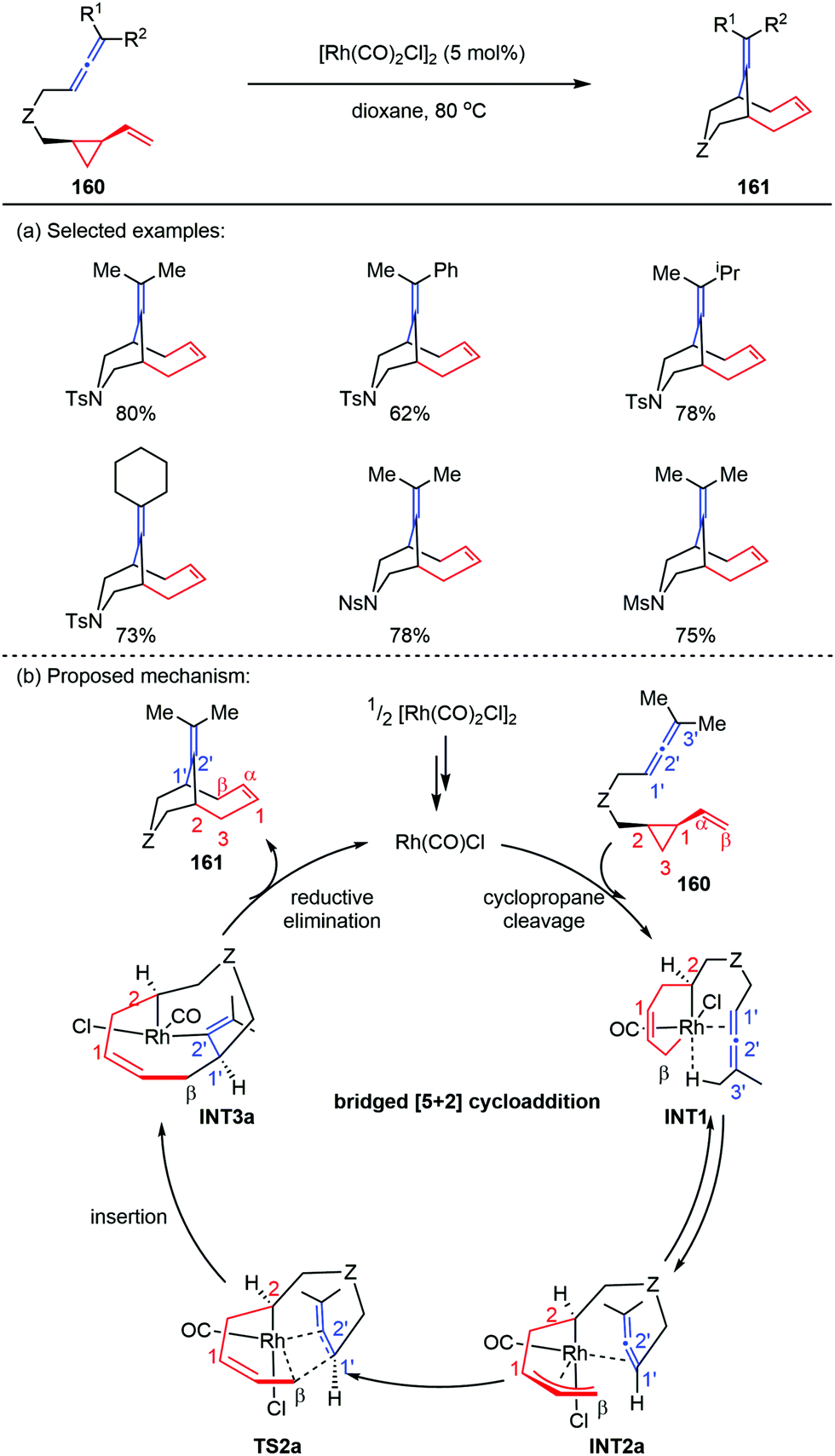
The previous report by Yu and co-workers showed that cis-ene-VCPs underwent a Rh-catalyzed [5+2] cycloaddition to provide 5,7-fused bicyclic products,76 where VCPs acted as five-carbon synthons. However, when they replaced the 2π component of cis-ene-VCPs with allene subunits, the corresponding cis-allene-VCPs 160 occurred an unprecedented bridged [5+2] cycloaddition to give the bicyclo[4.3.1]decane skeletons 161 (Scheme 28a).77 Preliminary DFT calculations revealed that the bridged [5+2] cycloaddition can reduce the repulsion effects by placing the allene moiety away from the VCP moiety in the allene insertion transition state (TS2a), thus affording the inversed allene insertion intermediate (INT3a). Finally, the reductive elimination led to the bridged cycloadducts 161 in good yields (Scheme 28b).
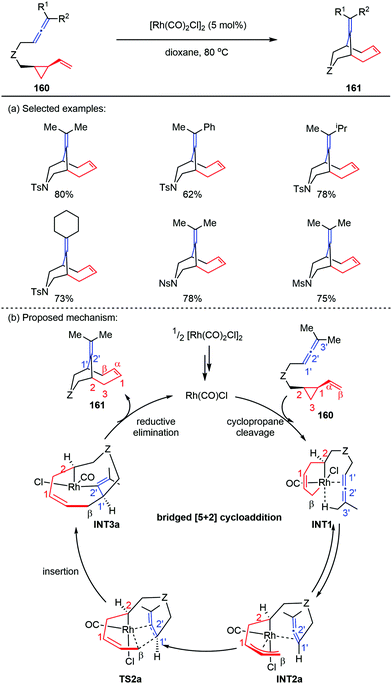 |
| | Scheme 28 [5+2] Cycloadditions of cis-allene-VCPs by Yu and co-workers. | |
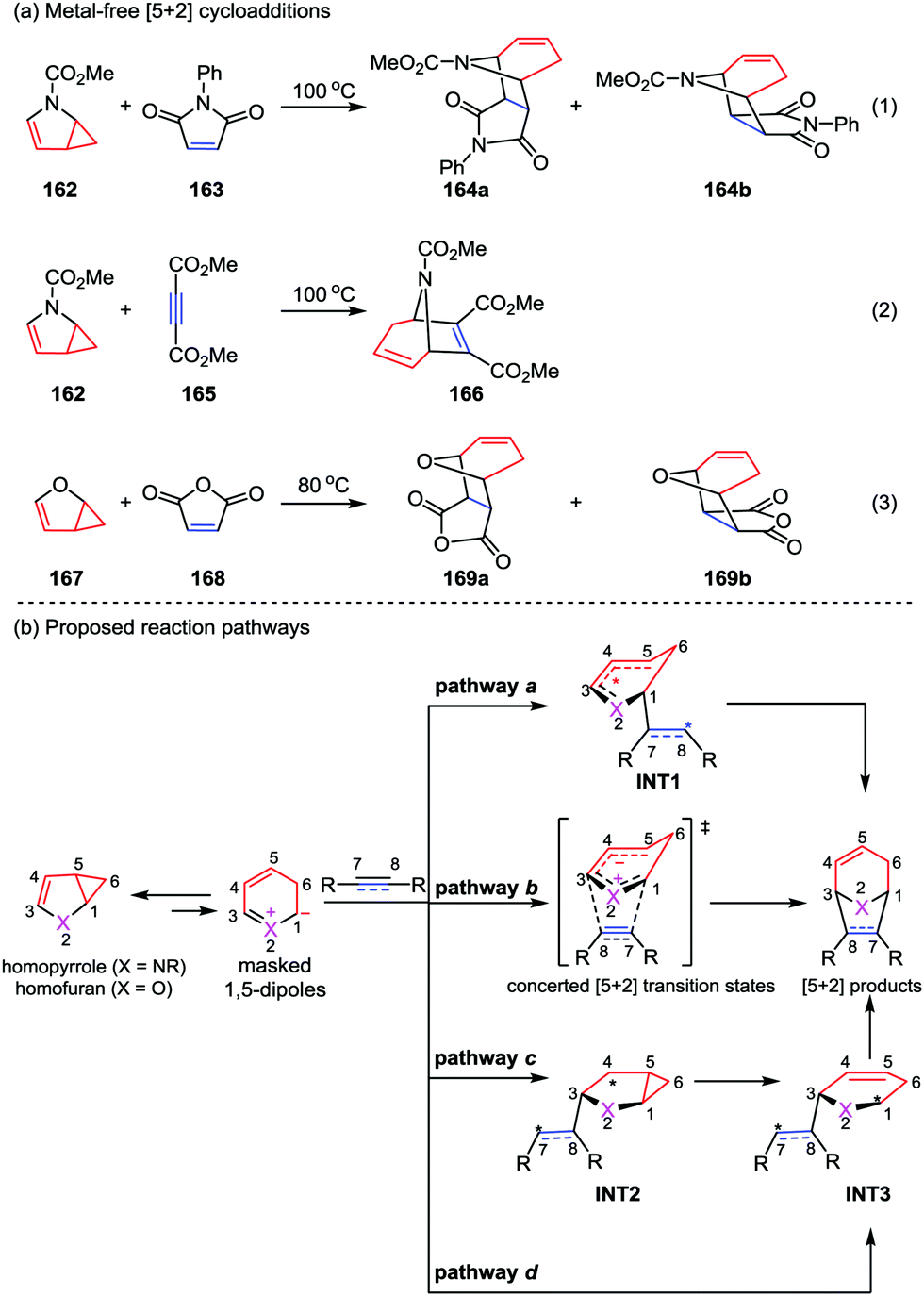
With the great progress of metal-catalyzed [5+2] cycloadditions between VCPs and alkenes/alkynes/allenes, mechanism studies on these reactions were successively reported by the group of Houk, Wender and Yu.78 Generally, these reactions occur through a series of elementary reactions including oxidative addition, alkene/alkyne/allene insertion, and reductive elimination. It is worth to note that recently the Yu group also contributed an important mechanism research work on the metal-free [5+2] reactions of N-methoxycarbonyl-2,3-homopyrrole 162 and homofuran 167 with dienophiles reported by Fowler (eqn (1) and (2)) and Ugi (eqn (3)) in 1971 and 1985, respectively (Scheme 29a).79 The [5+2] cycloaddition between N-methoxycarbonyl-2,3-homopyrrole 162 and N-phenylmaleimide 163 has been studied through the model reaction of N-methoxycarbonyl-2,3-homopyrrole and N-hydrogen-maleimide. DFT calculations of all four pathways a–d demonstrated that the reaction favors stepwise pathway a, giving diradical intermediate INT1, while in the concerted pathway b the cycloaddition between the existing-in-transition-state 1,5-dipole and dienophiles is disfavored. Moreover, although the a-exo is very similar to the a-endo pathway, it is disfavored by 1.6 kcal mol−1 in gas phase (1.7 kcal mol−1 in solution). These results indicate that the endo-cycloadduct is the favored one. In contrast, DFT calculations showed the [5+2] cycloaddition between homofuran 167 and maleic anhydride 168 prefers to occur concertedly via pathway b to give both the endo and exo [5+2] cycloadducts, while the competing diradical pathway a is marginally disfavored by 1 kcal mol−1. In addition, pathways c and d are not favored for both of the [5+2] cycloadditions studied, presumably due to the less resonance stabilization of the corresponding intermediates INT2 and INT3 compared with those in pathways a and b.79c
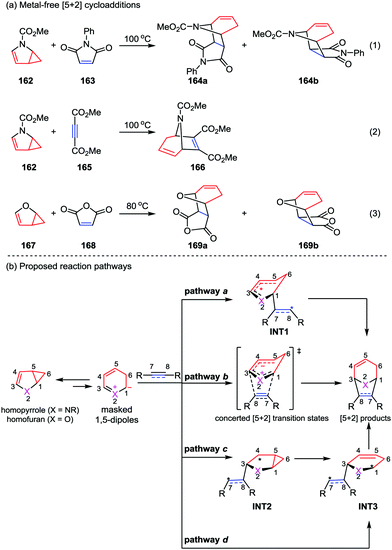 |
| | Scheme 29 (a) Metal-free [5+2] cycloadditions by Fowler and Ugi; (b) proposed concerted and stepwise pathways of the [5+2] cycloaddition by Yu and co-workers. | |
5.2 Hetero-[5+2] cycloaddition of vinyl aziridines and vinyl oxiranes
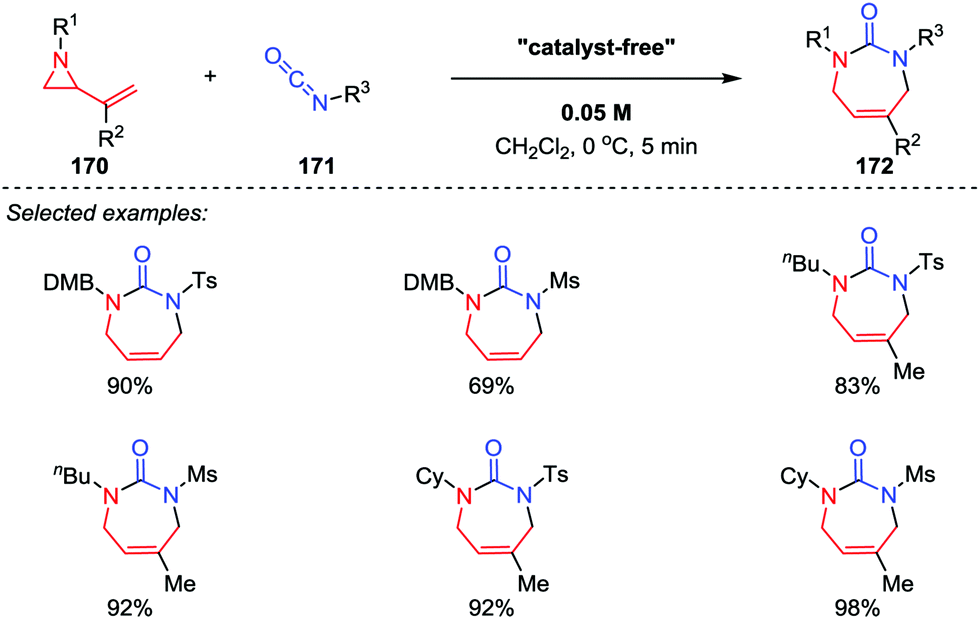
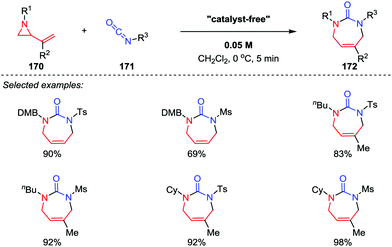
2-Vinyl aziridine is a strained and reactive cyclic amine, and a variety of reactions of vinyl aziridines involving the cleavage of the C–N bond have been reported.80 Among them, the intermolecular [5+2] cycloadditions of vinyl aziridines with unsaturated compounds represents one of the most efficient approaches for the synthesis of azepane frameworks. In 2012, Saito and co-workers demonstrated that the [5+2] cycloaddition reactions of 2-vinyl aziridines 170 with sulfonyl isocyanates 171 could take place without the aid of any catalysts, and the seven-membered cyclic ureas 172 were obtained in good to high yields (Scheme 30).81 Notably, the use of reactive sulfonyl isocyanates and the choice of dichloromethane as the solvent are two crucial factors for the proceeding of this reaction.
 |
| | Scheme 30 [5+2] Cycloadditions of vinyl aziridines and sulfonyl isocyanates by Saito and co-workers. | |
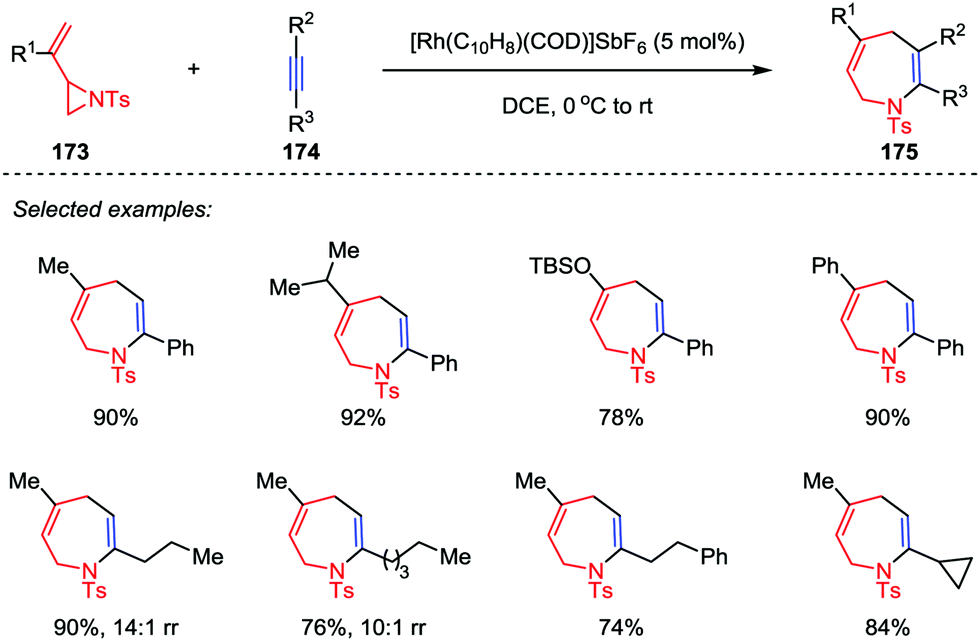
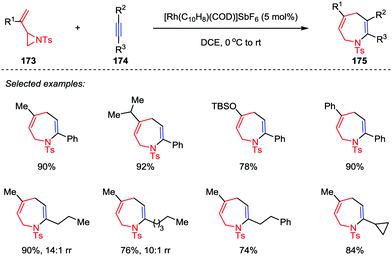
Although the intermolecular [3+2] cycloadditions of vinyl aziridines and activated alkenes have been explored and represent a powerful tool to obtain multi-substituted pyrrolidines, the more challenging intermolecular [5+2] cycloadditions of vinyl aziridines as five-atom components with general alkynes have rarely been explored. It was not until 2016, Zhang and co-workers disclosed the rhodium-catalyzed intermolecular [5+2] cycloaddition of vinyl aziridines 173 and alkynes 174 (Scheme 31).82 With the use of [Rh(C10H8)(COD)]SbF6 as catalyst, various aromatic and aliphatic terminal alkynes were converted to the corresponding cycloadducts 175 with high to excellent yields. In most cases, only one regioisomer was detected, except for some aliphatic alkynes (such as propyl and amyl substituted alkynes) affording the corresponding azepines 175 and their isomers in moderate to high ratios. Unfortunately, the internal alkynes were not applicable to this [5+2] cycloaddition.
 |
| | Scheme 31 Intermolecular [5+2] cycloadditions of vinyl aziridines and alkynes by Zhang and co-workers. | |
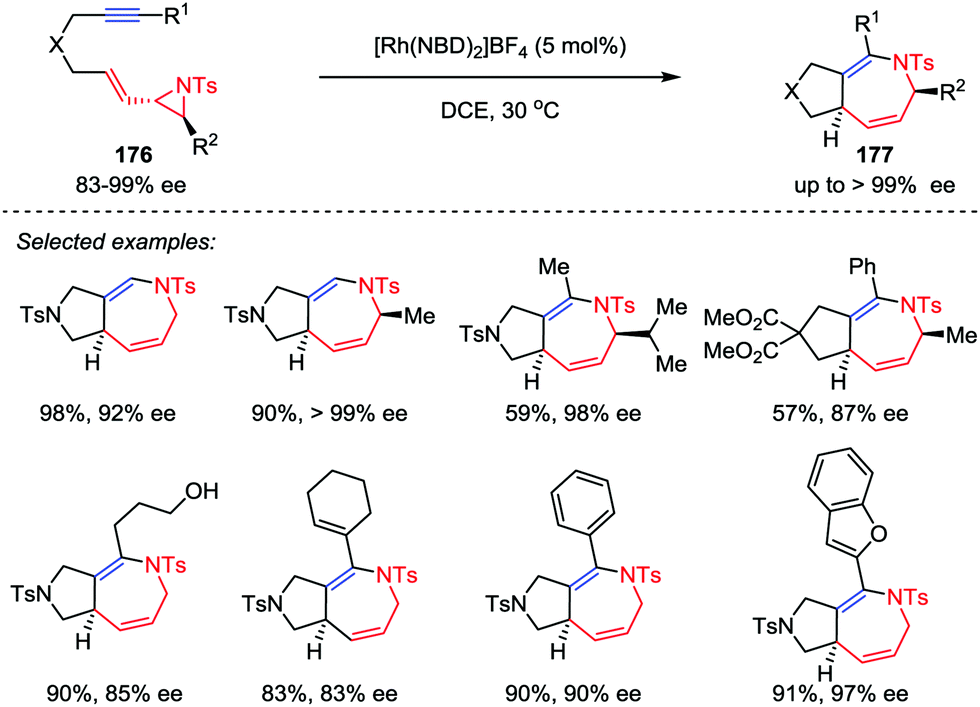
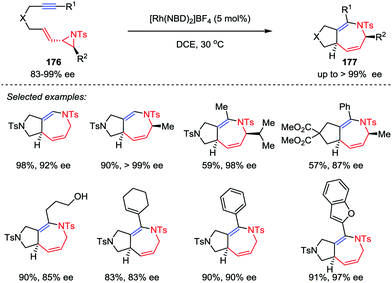
The development of new cycloaddition reactions to access polysubstituted and ring-fused azepines, especially the enantiomerically pure ones, from readily available acyclic precursors remains highly desirable in both organic and medicinal chemistry.83 Zhang and co-workers recently disclosed the stereoselective synthesis of fused 2,5-dihydroazepine derivatives via hetero-[5+2] cycloadditions of optically pure vinyl aziridine–alkyne substrates 176 in the presence of 5 mol% [Rh(NBD)2]BF4 catalyst (Scheme 32).84 Substrates with both internal and terminal alkyne moieties were compatible to afford the corresponding cycloaddition adducts 177 with high enantioselectivities. Moreover, this stereospecific cycloaddition reaction could be carried out on gram-scale with a reduced catalyst loading (2 mol% Rh).
 |
| | Scheme 32 Hetero-[5+2] cycloadditions of vinyl aziridines and alkynes by Zhang and co-workers. | |
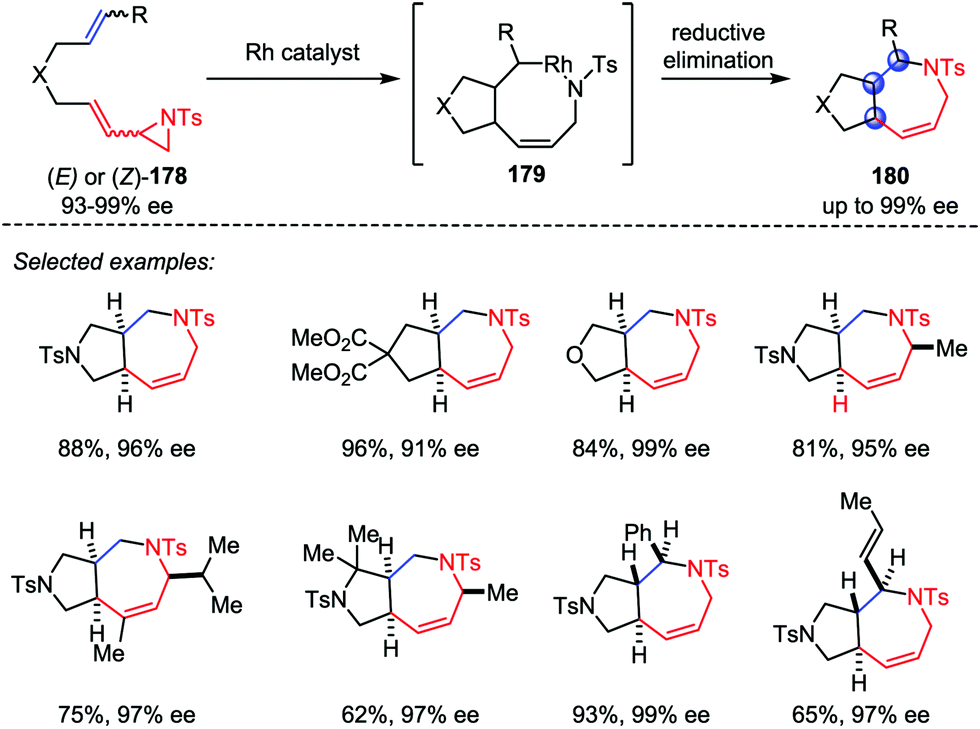
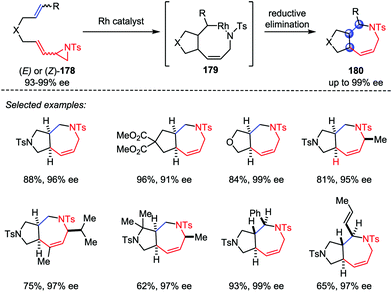
Based on the above work, Zhang and co-workers also developed a Rh-catalyzed intramolecular hetero-[5+2] cycloaddition of vinyl aziridines and alkenes (Scheme 33).85 Both internal and terminal alkenes were tolerated in this transformation. Various chiral-fused bicyclic azepines 180 with multiple contiguous stereogenic centers were obtained from the corresponding chiral starting materials 178 with good yields and high stereoselectivities under mild conditions.
 |
| | Scheme 33 Hetero-[5+2] cycloadditions of vinyl aziridines and alkenes by Zhang and co-workers. | |
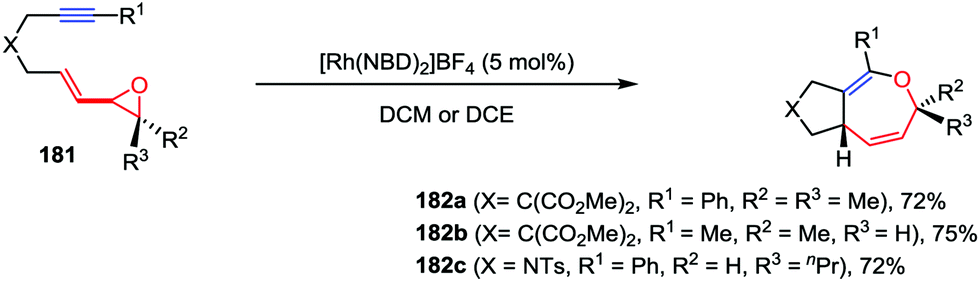
Oxepine derivatives represent a ubiquitous class of oxygen-containing seven-membered heterocycles with wide occurrence in many bioactive natural products and pharmaceuticals. Compared with the reactions that have been well established for five- and six-membered oxygen-containing heterocycles, the construction of oxepine skeletons is much more difficult due to the unfavorable entropic factors and transannular interactions.86 Owing to the inherent reactivity of their constrained ring systems, cyclopropane and oxirane derivatives constitute valuable building blocks for organic synthesis. In contrast to VCPs, vinyloxiranes and their derivatives have served as attractive ring-expansion motifs in metal-catalyzed [3+2] cycloaddition, epoxide carbonylation and substitution reactions.87 Recently, Zhang and co-workers described an intramolecular hetero-[5+2] cycloaddition of vinyloxirane-alkynes 181 in the presence of 5 mol% of [Rh(NBD)2]BF4, affording the corresponding bicyclic 2,5-dihydrooxepine derivatives 182 in high yields (Scheme 34).88
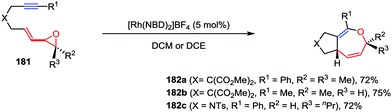 |
| | Scheme 34 Intramolecular [5+2] cycloadditions of vinyl oxiranes and internal alkynes by Zhang and co-workers. | |
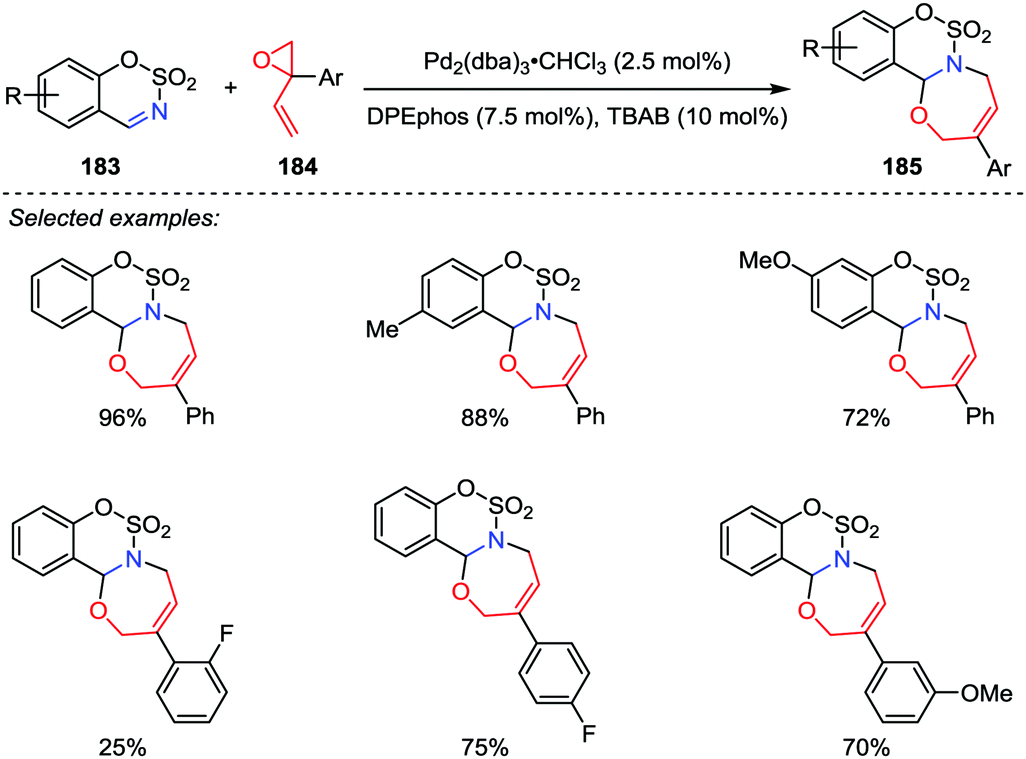
1,3-Oxazepines are seven-membered heterocyclic compounds bearing both nitrogen and oxygen atoms in the ring, which have been extensively studied due to their potent biological activities. Although a considerable number of ways for the synthesis of 1,3-oxazepine ring have been reported,89 most of these methods suffered from the drawbacks such as substrate preparation, multiple-step reactions and low yields. Therefore, novel and efficient protocols for the oxazepine ring construction are highly desirable. In 2017, Guo and co-workers reported a palladium-catalyzed [5+2] cycloaddition of sulfamate-derived cyclic imines 183 with 2-aryl-2-vinyloxiranes 184, affording biologically interesting sulfamate-fused 1,3-oxazepine derivatives 185 in good to excellent yields with satisfactory regioselectivities (Scheme 35).90 However, the unsubstituted vinyloxiranes and alkyl-substituted vinyloxiranes were found unable to undergo this reaction.
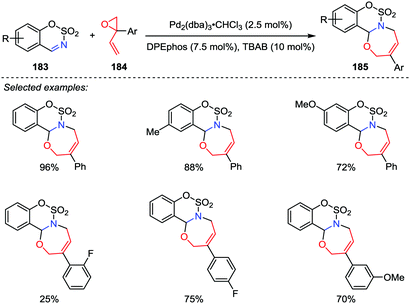 |
| | Scheme 35 [5+2] Cycloadditions of vinyl oxiranes and cyclic imines by Guo and co-workers. | |
5.3 Natural product synthesis
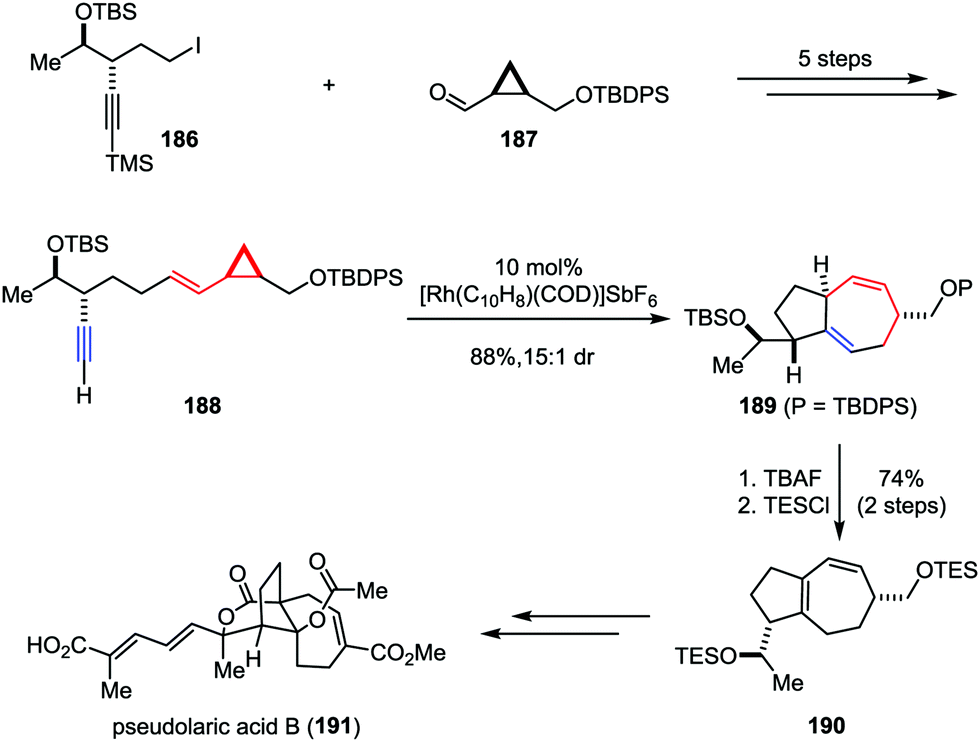
Pseudolaric acid B, a diterpene acid, was isolated from the bark of Pseudolarix kaempferi Gordon (pinaceae).91 It has been identified as a potent antifungal, antifertility, and cytotoxic agent.92 Furthermore, pseudolaric acid B inhibits angiogenesis by diminishing the secretion of vascular endothelial growth factor (VEGF) in tumor cells and has been shown to induce apoptosis through several pathways in human melanoma cells.93 The remarkable biological properties and the low toxicity of pseudolaric acid B have rendered it an attractive target for synthetic studies. However, the unusual trans-fused [5,7] bicyclic ring system (polyhydroazulene) makes the stereoselective approach toward this molecule a daunting task. In 2007, Trost and co-workers described the total synthesis of pseudolaric acid B (Scheme 36).94 Their synthesis began with the preparation of linear precursor 188, which could be derived from iodide 186 and aldehyde 187 in five steps. The planned Rh-catalyzed intramolecular [5+2] cycloaddition of 188 proceeded smoothly to construct the polyhydroazulene core, providing the desired product 189 in 88% yield with 15:1 dr. Subsequently, the TBAF-induced isomerization of 1,4-diene to 1,3-diene followed by resilylation afforded 190 in 74% yield over two steps. Further elaboration of 190 led to pseudolaric acid B (191).
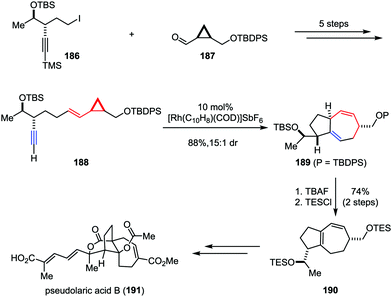 |
| | Scheme 36 Total synthesis of (−)-pseudolaric acid B by Trost and co-workers. | |
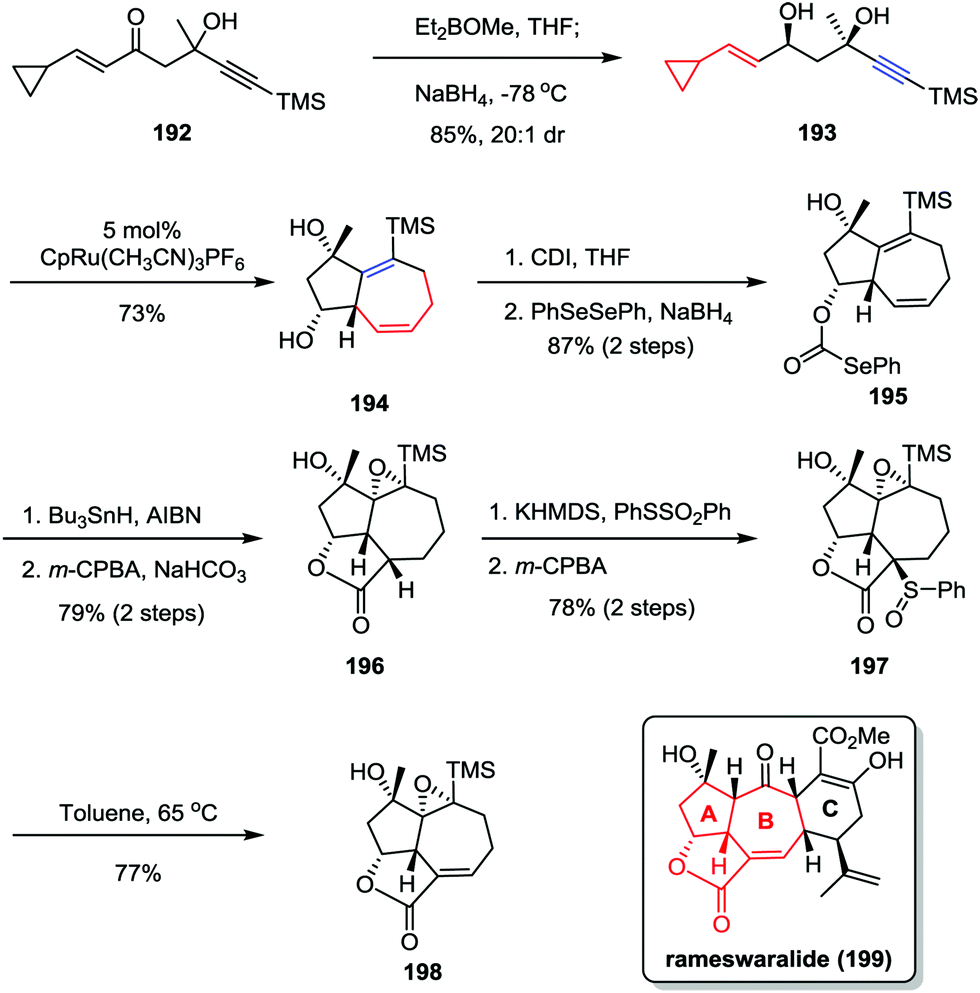
Rameswaralide, a novel diterpene with a tetracyclic ring skeleton, was isolated from the soft coral Sinularia dissecta near the Mandapam coast of India in 1998.95 Rameswaralide and its derivatives could function as effective anti-inflammatory agents for the treatment of a variety of inflammatory disorders, including arthritis, psoriasis, and inflammatory bowel disease.96 In 2011, the Trost group reported an efficient synthetic approach toward the tricyclic structure containing A/B rings of rameswaralide (199), which featured the formation of the challenging [5,7] bicyclic skeleton and the exocyclic α,β-unsaturated lactone subunit (Scheme 37).97 After the preparation of syn-1,3-diol 193 through a diastereoselective 1,2-reduction of enone 192, the Ru-catalyzed intramolecular [5+2] cycloaddition resulted in the formation of the [5,7] bicycle 194 as a single isomer in good yield. Subsequent reductive radical cyclization followed by oxidation/regioselective elimination afforded the desired lactone 198 smoothly.
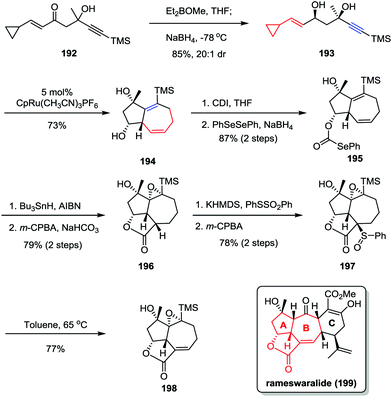 |
| | Scheme 37 Synthesis of the tricyclic core of rameswaralide by Trost and co-workers. | |
6. [5+2] Cycloadditions of 5-carbon organo-metallic complexes
6.1 [5+2] Cycloadditions of 5-carbon cobalt and molybdenum complexes
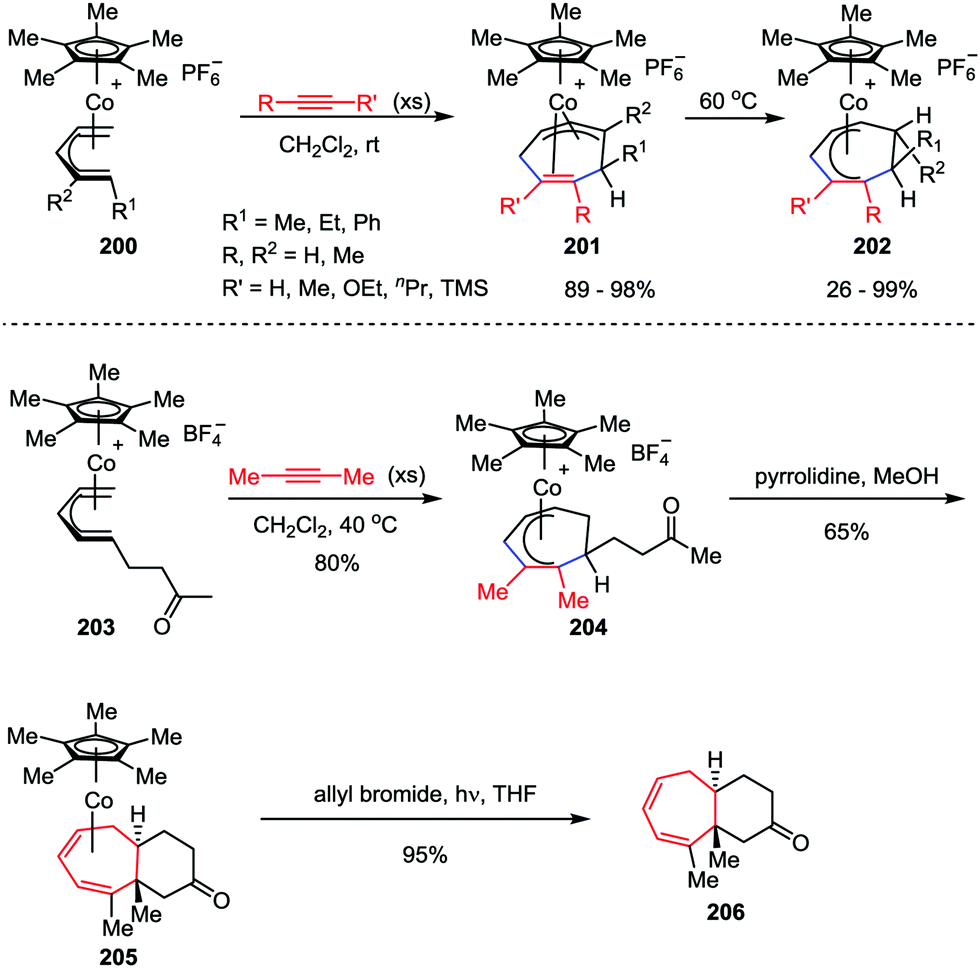
Novel transition-metal-mediated reactivity patterns also provided the conceptual basis for the development of new synthetic reactions. Despite considerable advancements having been made, the [5+2] cycloaddition reaction of an alkyne and η5-pentadienyl complex still remains an attractive topic for organic chemists. In 2008, Stryker and co-workers discovered an interesting [5+2] cycloaddition of acyclic (open) η5-pentadienyl cobalt complexes 200 and alkynes under notably mild conditions (Scheme 38).98 The reaction selectively afforded the cycloheptadienyl ring as the η2,η3-coordinated isomer 201, which was an unusual coordination mode for unbridged cycloheptadienyl systems. Through gentle heating, the isomer could be transformed to the fully conjugated η5-cycloheptadienyl product 202. They further investigated this reaction manifold by extending the reaction to tandem cycloaddition/nucleophilic cyclization to generate fused bicyclic compounds.99 The cycloaddition of η5-pentadienyl ketone complex 203 with high concentrations of 2-butyne afforded η5-cycloheptadienyl ketone complex 204. Upon exposure of 204 to pyrrolidine, the expected bicyclic complex 205 was obtained as a single diastereoisomer in a reasonable yield. The subsequent photolytic decomplexation proceeded smoothly, delivered bicyclic ketone 206 with 95% isolated yield. Gratifyingly, no loss of stereochemical or compositional integrity was detected (Scheme 38).
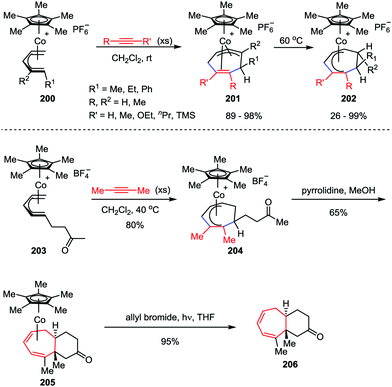 |
| | Scheme 38 Cobalt-mediated η5-pentadienyl/alkyne [5+2] cycloaddition by Stryker and co-workers. | |
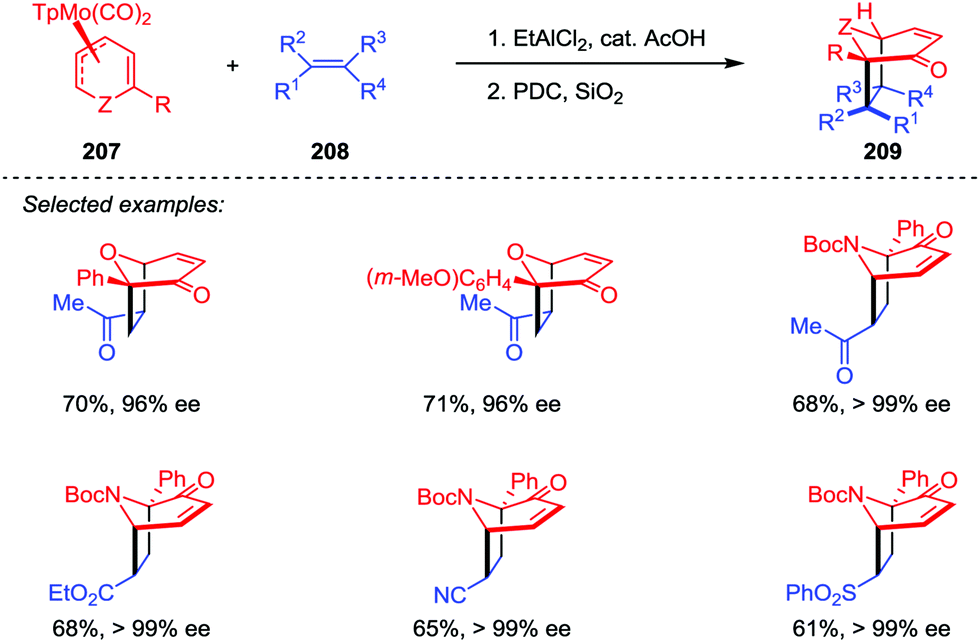
The oxa- and azabicyclo[3.2.1]octene skeletons are key structural subunits existing in a large and diverse array of biologically important natural products. Although their synthesis through [5+2] cycloadditions of 3-oxidopyrylium and 3-oxidopyridinium dipoles has been extensively studied, these approaches often suffered from drawbacks such as the limited methods in asymmetric version and the lack of facial- and regioselective control in cycloaddition. An Intriguing progress was reported by Liebeskind and co-workers.100 As shown in Scheme 39, a series of 6-substituted η3-pyranyl- and η3-pyridinylmolybdenum scaffolds 207 have been prepared, which were then applied in a novel Brønsted acid-catalyzed [5+2] cycloaddition and served as chiral substrates reacting with a range of electron-deficient alkenes 208. After the oxidative demetalation by using PDC/SiO2, a variety of 2-substituted oxa- and azabicyclo[3.2.1]octene rings 209 were obtained with complete retention of high enantioselectivities. Moreover, this reaction enabled the formation of four stereogenic centers in one step, as well as the control of both regio- and stereoselectivities.
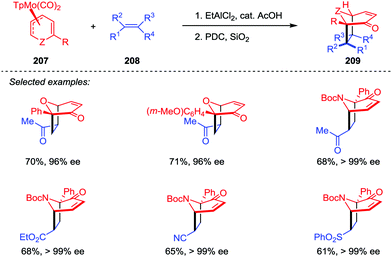 |
| | Scheme 39 [5+2] Cycloaddition of η3-pyranyl- and η3-pyridinylmolybdenum complexes and alkenes by Liebeskind and co-workers. | |
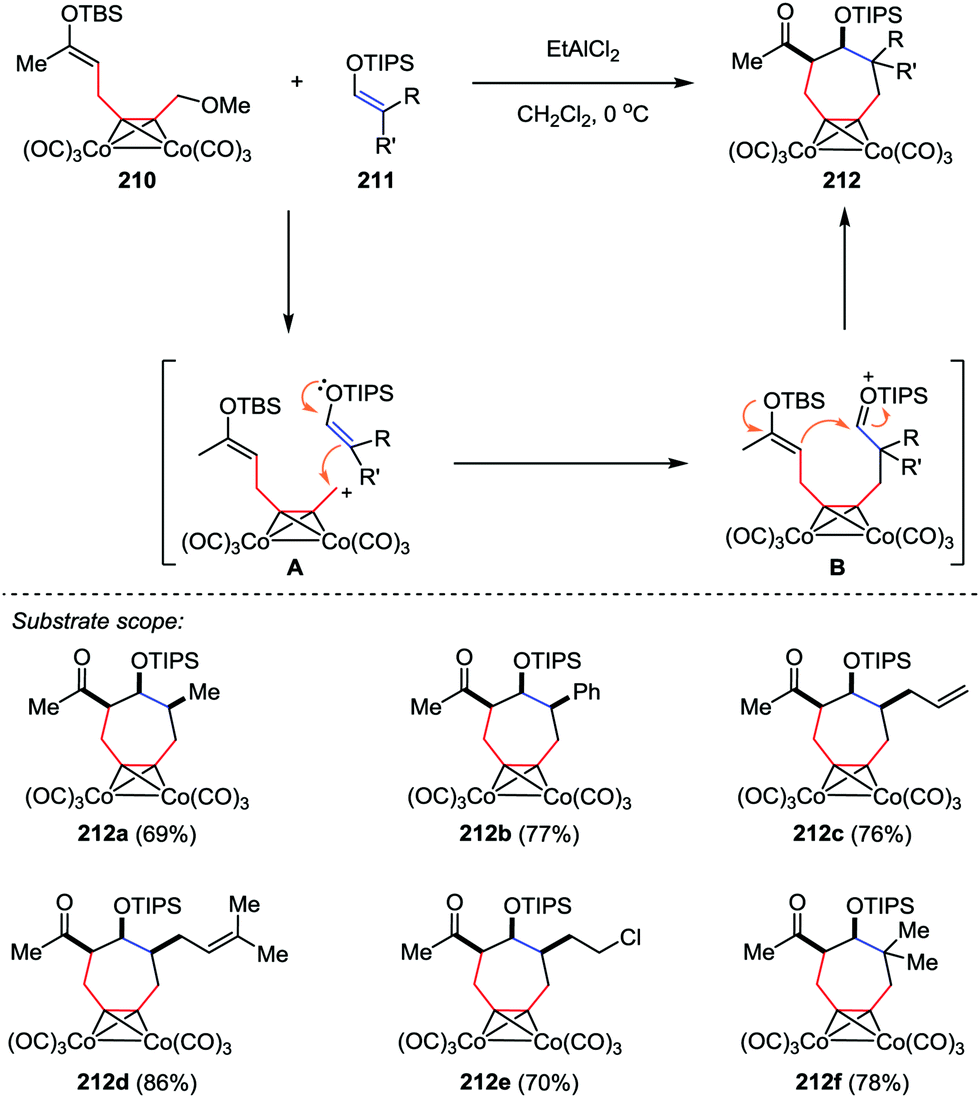
The cycloheptane ring bearing various kinds of substituents is a common skeleton in a number of natural products. Efficient methods have been reported for the construction of these seven-membered carbocycles through [4+3] cycloaddition of dienes or furan derivatives and allylic cationic species.101 However, much less attention was paid to the [5+2] cycloaddition. In 2014, Tanino and co-workers reported a [5+2] cycloaddition of dicobalt acetylene complexes 210 and enol silyl ethers 211 (Scheme 40).102 This reaction proceeded stepwisely through intermolecular addition of cationic species A with an enol silyl ether followed by the intramolecular cyclization of silyloxonium ion B in the presence of EtAlCl2, affording the cycloheptanes 212 in good yields with high stereoselectivities. Further structural analysis revealed that three substituents on the cycloheptane ring of 212a–e are all cis to each other.
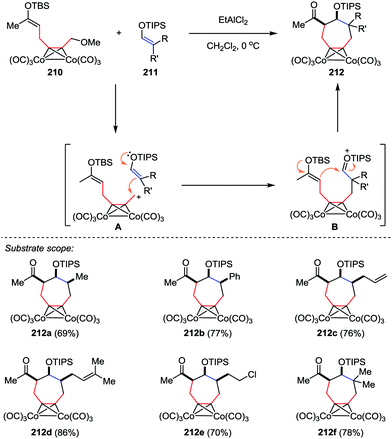 |
| | Scheme 40 [5+2] Cycloaddition reactions of dicobalt acetylene complexes with enol silyl ethers by Tanino and co-workers. | |
6.2 Natural product synthesis
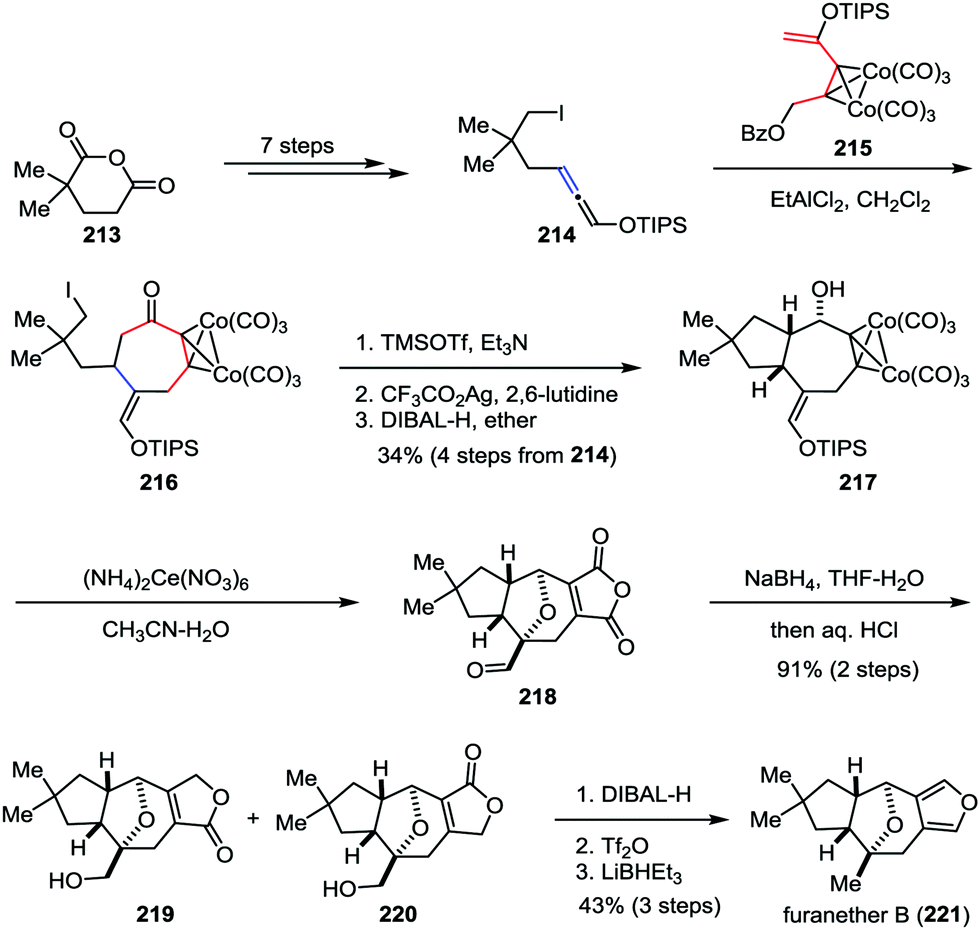
Recently, Tanino and co-workers developed a novel [5+2] cycloaddition reaction between a dicobalt hexacarbonyl propargyl cation species and a silyloxyallene for the formation of functionalized cycloheptanones.103 This methodology was subsequently applied as a key step in the total synthesis of furanether B (Scheme 41). Their synthesis commenced with the preparation of allene 214, which was derived from commercially available 2,2-dimethylglutaric anhydride 213 in seven steps. An intermolecular [5+2] cycloaddition reaction between allene 214 and cobalt complex 215 afforded the desired cycloheptanone 216. After the cyclopentane formation and stereoselective 1,2-reduction of the ketone subunit, bicyclic alcohol 217 was formed in 34% yield over four steps. Upon treatment of 217 with an excess amount of cerium(IV) ammonium nitrate led to the formation of aldehyde 218. The results indicate that the secondary hydroxy group of the substrate underwent transannular oxidative coupling with the enol silyl ether moiety, which allowed the total synthesis to finish more quickly. NaBH4 reduction of aldehyde 218 followed by acidic workup gave rise to hydroxy lactone 219 as a mixture with its regioisomer 220 in high yield. Finally, furanether B (221) was furnished through further reduction.
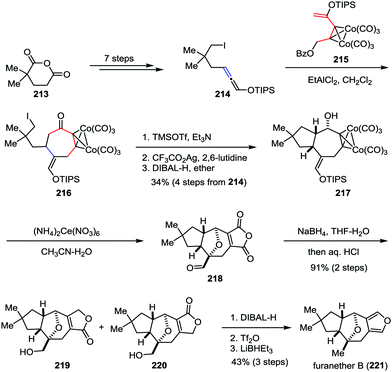 |
| | Scheme 41 Total synthesis of furanether B by Tanino and co-workers. | |
7. Conclusions and outlook
This review summarized the recent advances in [5+2] cycloadditions, especially on instructive and fascinating applications in natural product synthesis. The rapid generation of molecular complexity in a relatively concise manner has rendered the [5+2] cycloaddition protocol a highly useful tool, which were highlighted with the latest progress in various reaction intermediates involved, such as oxidopyrylium ylides, oxidopyridinium ylides, perezone-type intermediates, vinylsubstituted three-membered rings and 5-carbon organometallic complexes. As demonstrated in these examples, the steric effects of the tethered side chains or chiral auxiliaries are vital for the retention of the geometry of the active ions. On the other hand, biomimetic syntheses have proven to be promising protocols for the construction of highly complex fused seven-membered ring skeletons, and the efficiencies on the ODI-[5+2] cycloaddition cascade approach to forge the complex polycyclic core structures have also been demonstrated.
With the continuing attention paid to this area, we believe more effective methods and strategies for the development of new types of [5+2] cycloadditions would certainly emerge and be successfully applied in the total synthesis of more natural products with biological importance.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We acknowledge the financial support from Zhejiang Natural Science Fund for Distinguished Young Scholars (LR16B020001) and National Natural Science Foundation of China (21871230, 21622205).
References
-
(a) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS PubMed;
(b) M. Harmata, Chem. Commun., 2010, 46, 8904–8922 RSC;
(c) M. A. Casadei, C. Galli and L. Mandolini, J. Am. Chem. Soc., 1984, 106, 1051–1056 CrossRef CAS;
(d) G. Illuminati and L. Mandolini, Acc. Chem. Res., 1981, 14, 95–102 CrossRef CAS;
(e) A. Parenty, X. Moreau and J.-M. Campagne, Chem. Rev., 2006, 106, 911–939 CrossRef CAS PubMed.
-
(a) P. Li and D. Menche, Angew. Chem., Int. Ed., 2009, 48, 5078–5080 CrossRef CAS PubMed;
(b) A. G. Lohse and R. P. Hsung, Chem. – Eur. J., 2011, 17, 3812–3822 CrossRef CAS PubMed;
(c) M. A. Battiste, P. M. Pelphrey and D. L. Wright, Chem. – Eur. J., 2006, 12, 3438–3447 CrossRef CAS PubMed.
- W. Leather and R. Anschütz, Chem. Ber., 1885, 18, 715–717 CrossRef.
-
(a) D. Zhao, J. Zhang and Z. Xie, J. Am. Chem. Soc., 2015, 137, 13938–13942 CrossRef CAS PubMed;
(b) C. Hu, R.-J. Song, M. Hu, Y. Yang, J.-H. Li and S. Luo, Angew. Chem., Int. Ed., 2016, 55, 10423–10426 CrossRef CAS PubMed.
-
(a) K. L. Cubbage, A. J. Orr-Ewing and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2009, 48, 2514–2517 CrossRef CAS PubMed;
(b) B. D. A. Hook, W. Dohle, P. R. Hirst, M. Pickworth, M. B. Berry and K. I. Booker-Milburn, J. Org. Chem., 2005, 70, 7558–7564 CrossRef CAS PubMed.
- K. E. O. Ylijoki and J. M. Stryker, Chem. Rev., 2013, 113, 2244–2266 CrossRef CAS PubMed.
-
(a) H. Pellissier, Adv. Synth. Catal., 2011, 353, 189–218 CrossRef CAS;
(b) H. Pellissier, Adv. Synth. Catal., 2018, 360, 1551–1583 CrossRef CAS.
- X. Liu, Y.-J. Hu, J.-H. Fan, J. Zhao, S. Li and C.-C. Li, Org. Chem. Front., 2018, 5, 1217–1228 RSC.
- L. P. Bejcek and R. P. Murelli, Tetrahedron, 2018, 74, 2501–2521 CrossRef CAS PubMed.
-
(a) X. Shu, S. Huang, D. Shu, I. A. Guzei and W. Tang, Angew. Chem., Int. Ed., 2011, 50, 8153–8156 CrossRef CAS PubMed;
(b) X.-Z. Shu, X. Li, D. Shu, C. M. Schienebeck, X. Zhou, P. J. Robichaux and W. Tang, J. Am. Chem. Soc., 2004, 126, 16300–16301 CrossRef PubMed;
(c) X. Shu, C. M. Schienebeck, W. Song, I. A. Guzei and W. Tang, Angew. Chem., Int. Ed., 2013, 52, 13601–13605 CrossRef CAS PubMed;
(d) X. Li, W. Song, X. Ke, X. Xu, P. Liu, K. N. Houk, X. Zhao and W. Tang, Chem. – Eur. J., 2016, 22, 7079–7083 CrossRef CAS PubMed.
-
(a) A. Seoane, N. Casanova, N. Quiñones, J. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2014, 136, 834–837 CrossRef CAS PubMed;
(b) R. Shenje, M. C. Martin and S. France, Angew. Chem., Int. Ed., 2014, 53, 13907–13911 CrossRef CAS PubMed;
(c) L. Bai, Y. Wang, Y. Ge, J. Liu and X. Luan, Org. Lett., 2017, 19, 1734–1737 CrossRef CAS PubMed;
(d) N. Casanova, K. P. Del Rio, R. García-Fandiño, J. L. Mascareñas and M. Gulías, ACS Catal., 2016, 6, 3349–3353 CrossRef CAS PubMed;
(e) B. Cendón, N. Casanova, C. Comanescu, R. García-Fandiño, A. Seoane, M. Gulías and J. L. Mascareñas, Org. Lett., 2017, 19, 1674–1677 CrossRef PubMed.
-
(a) I. Kuwajima and K. Tanino, Chem. Rev., 2005, 105, 4661–4670 CrossRef CAS PubMed;
(b) S. Kim and J. Winkler, Chem. Soc. Rev., 1997, 26, 387–399 RSC;
(c) L. E. Brown and J. P. Konopelski, Org. Prep. Proced. Int., 2008, 40, 411–445 CrossRef CAS.
-
(a) A. Nickel, T. Maruyama, H. Tang, P. D. Murphy, B. Greene, N. Yusuff and J. L. Wood, J. Am. Chem. Soc., 2004, 126, 16300–16301 CrossRef CAS PubMed;
(b) I. Kuwajima and K. Tanino, Chem. Rev., 2005, 105, 4661–4670 CrossRef CAS PubMed.
- C. Plummer, C. S. Wei, C. E. Yozwiak, A. Soheili, S. O. Smithback and J. L. Leighton, J. Am. Chem. Soc., 2014, 136, 9878–9881 CrossRef CAS PubMed.
- G. Mei, X. Liu, C. Qiao, W. Chen and C.-C. Li, Angew. Chem., Int. Ed., 2015, 54, 1754–1758 CrossRef CAS PubMed.
-
(a) P. A. Wender and J. L. Mascareñas, J. Org. Chem., 1991, 56, 6267–6269 CrossRef CAS;
(b) P. A. Wender and J. L. Mascareñas, Tetrahedron Lett., 1992, 33, 2115–2118 CrossRef CAS.
- G. Mei, H. Yuan, Y. Gu, W. Chen, L. W. Chung and C.-C. Li, Angew. Chem., Int. Ed., 2014, 126, 11231–11235 CrossRef.
- R. H. Kaufman, C. M. Law, J. A. Simanis, E. L. Woodall, C. R. Zwick, III, H. B. Wedler, P. Wendelboe, C. G. Hamaker, J. R. Goodell, D. J. Tantillo and T. A. Mitchell, J. Org. Chem., 2018, 83, 9818–9838 CrossRef CAS PubMed.
-
(a) N. Z. Burns, M. R. Witten and E. N. Jacobsen, J. Am. Chem. Soc., 2011, 133, 14578–14581 CrossRef CAS PubMed;
(b) M. R. Witten and E. N. Jacobsen, Angew. Chem., Int. Ed., 2014, 53, 5912–5916 CrossRef CAS PubMed.
- A. Orue, U. Uria, E. Reyes, L. Carrillo and J. L. Vicario, Angew. Chem., Int. Ed., 2015, 54, 3043–3046 CrossRef CAS PubMed.
-
(a) R. A. Volkmann, P. D. Weeks, D. E. Kuhla, E. B. Whipple and G. N. Chmurny, J. Org. Chem., 1977, 42, 3976–3978 CrossRef CAS;
(b) M. E. Garst, B. J. McBride and J. Douglass, Tetrahedron Lett., 1983, 24, 1675–1678 CrossRef CAS;
(c) P. A. Wender and F. E. McDonald, J. Am. Chem. Soc., 1990, 112, 4956–4958 CrossRef CAS;
(d) A. Rumbo, A. Mouriño, L. Castedo and J. L. Mascareñas, J. Org. Chem., 1996, 61, 6114–6120 CrossRef CAS PubMed;
(e) J. L. Mascareñas, I. Perez, A. Rumbo and L. Castedo, Synlett, 1997, 81–82 Search PubMed;
(f) J. R. Rodrıguez, A. Rumbo, L. Castedo and J. L. Mascareñas, J. Org. Chem., 1999, 64, 4560–4563 CrossRef;
(g) P. A. Wender, N. D’ Angelo, V. I. Elitzin, M. Ernst, E. E. Jackson-Ugueto, J. A. Kowalski, S. McKendry, M. Rehfeuter, R. Sun and D. Voigtlaender, Org. Lett., 2007, 9, 1829–1832 CrossRef CAS PubMed.
-
(a) M. P. D’Erasmo, C. Meck, C. A. Lewis and R. P. Murelli, J. Org. Chem., 2016, 81, 3744–3751 CrossRef;
(b) K. N. Fuhr, D. R. Hirsch, R. P. Murelli and S. E. Brenner-Moyer, Org. Lett., 2017, 19, 6356–6359 CrossRef CAS.
- H. Suga, T. Iwai, M. Shimizu, K. Takahashi and Y. Toda, Chem. Commun., 2018, 54, 1109–1112 RSC.
- B. B. Snider, X. Wu, S. Nakamura and S. Hashimoto, Org. Lett., 2007, 9, 873–874 CrossRef CAS PubMed.
- S. Nakamura, Y. Sugano, F. Kikuchi and S. Hashimoto, Angew. Chem., Int. Ed., 2006, 45, 6532–6535 CrossRef CAS PubMed.
- R. Ratnayake, D. Covell, T. T. Ransom, K. R. Gustafson and J. A. Beutler, Org. Lett., 2009, 11, 57–60 CrossRef CAS PubMed.
- K. C. Nicolaou, Q. Kang, S. Y. Ng and D. Y.-K. Chen, J. Am. Chem. Soc., 2010, 132, 8219–8222 CrossRef CAS PubMed.
- J. G. Buta, J. L. Flippen and W. R. Lusby, J. Org. Chem., 1978, 43, 1002–1003 CrossRef CAS.
- N.-J. Sun, Z. Xue, X.-T. Liang and L. Huang, Acta Pharmacol. Sin., 1979, 14, 39–43 CAS.
- S. Q. Kang, S. Y. Cai and L. Teng, Acta Pharmacol. Sin., 1981, 16, 867–868 CAS.
- M. Zhang, N. Liu and W. Tang, J. Am. Chem. Soc., 2013, 135, 12434–12438 CrossRef CAS PubMed.
- J. Kinjo, H. Uemura, T. Nohara, M. Yamashita, N. Marubayashi and K. Yoshihira, Tetrahedron Lett., 1995, 36, 5599–5602 CrossRef CAS.
- S. Strych, G. Journot, R. P. Pemberton, S. C. Wang, D. J. Tantillo and D. Trauner, Angew. Chem., Int. Ed., 2015, 54, 5079–5083 CrossRef CAS PubMed.
- S. Strych and D. Trauner, Angew. Chem., Int. Ed., 2013, 52, 9509–9512 CrossRef CAS PubMed.
-
(a) M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed;
(b) S. Lachkar, A. Sobel and M. Knossow, Nature, 2004, 428, 198–202 CrossRef PubMed.
- J. L. Weiner, A. V. Buhler, V. J. Whatley, R. A. Harris and T. V. Dunwiddie, J. Pharmacol. Exp. Ther., 1998, 284, 95–102 CAS.
- B. Chen, X. Liu, Y.-J. Hu, D.-M. Zhang, L. Deng, J. Lu, L. Min, W.-C. Ye and C.-C. Li, Chem. Sci., 2017, 8, 4961–4966 RSC.
- J. X. Zhao, C. P. Liu, W. Y. Qi, M. L. Han, Y. S. Han, M. A. Wainberg and J. M. Yue, J. Nat. Prod., 2014, 77, 2224–2233 CrossRef CAS PubMed.
- X. Liu, J. Liu, J. Zhao, S. Li and C.-C. Li, Org. Lett., 2017, 19, 2742–2745 CrossRef CAS PubMed.
- L. Du, T. Zhu, Y. Fang, Q. Gu and W. Zhu, J. Nat. Prod., 2008, 71, 1343–1351 CrossRef CAS PubMed.
- J. Liu, J. Wu, J.-H. Fan, X. Yan, G. Mei and C.-C. Li, J. Am. Chem. Soc., 2018, 140, 5365–5369 CrossRef CAS PubMed.
- P. A. Wender, T. M. Pedersen and M. J. C. Scanio, J. Am. Chem. Soc., 2002, 124, 15154–15155 CrossRef CAS PubMed.
- D. J. Lee, H. S. Han, J. Shin and E. J. Yoo, J. Am. Chem. Soc., 2014, 136, 11606–11609 CrossRef CAS PubMed.
- N. De, C. E. Song, D. H. Ryu and E. J. Yoo, Chem. Commun., 2018, 54, 6911–6914 RSC.
-
(a) S.-H. Lim, Y.-Y. Low, S. K. Sinniach, K.-T. Yong, K.-S. Sim and T.-S. Kam, Phytochemistry, 2014, 98, 204–215 CrossRef CAS PubMed;
(b) M. Lounasmaa, P. Hanhinen and M. Westersund, Alkaloids, 1999, 52, 103–195 CAS.
- S. Krüger and T. Gaich, Angew. Chem., Int. Ed., 2015, 54, 315–317 CrossRef PubMed.
-
(a) A. R. Katritzky and N. Dennis, Chem. Rev., 1989, 89, 827–861 CrossRef CAS;
(b) V. K. Aggarwal, J. Drabowicz, R. S. Grainger, Z. Gultekin, M. Lightowler and P. L. Spargo, J. Org. Chem., 1995, 60, 4962–4963 CrossRef CAS.
- J. C. Green and T. R. R. Pettus, J. Am. Chem. Soc., 2011, 133, 1603–1608 CrossRef CAS PubMed.
- C. He, J. Hu, Y. Hu and H. Ding, J. Am. Chem. Soc., 2017, 139, 6098–6101 CrossRef CAS PubMed.
- C. He, Z. Bai, J. Hu, B. Wang, H. Xie, L. Yu and H. Ding, Chem. Commun., 2017, 53, 8435–8438 RSC.
-
(a) F.-P. Wang and L.-P. Yan, Tetrahedron, 2007, 63, 1417–1420 CrossRef CAS;
(b) D. L. Selwood, S. R. Challand, J. N. Champness, J. Gillmam, D. K. Hibberd, K. S. Jandu, D. Lowe, M. Pether, J. Selway and G. E. Trantor, J. Med. Chem., 1993, 36, 3503–3510 CrossRef CAS PubMed;
(c) R. H. Guo, C. X. Guo, D. He, D. K. Zhao and Y. Shen, Chin. J. Chem., 2017, 35, 1644–1647 CrossRef CAS.
- Y. Liu, X. Wang, S. Chen, S. Fu and B. Liu, Org. Lett., 2018, 20, 2934–2938 CrossRef CAS PubMed.
-
(a) P. Walter, Annalen, 1841, 39, 244–247 CrossRef;
(b) P. Walter, Annalen, 1843, 48, 35–38 CrossRef.
- J. Oda, N. Ando, Y. Nakajima and Y. Inouye, Agric. Biol. Chem., 1977, 41, 201–204 CAS.
- R. P. Adams, C. A. McDaniel and F. L. Carter, Biochem. Syst. Ecol., 1988, 16, 453–456 CrossRef.
- F. Walls, P. Padilla, P. Joseph-Nathan, F. Giral and J. Romo, Tetrahedron Lett., 1965, 21, 1577–1582 CrossRef.
- H. Takigawa, H. Kubota, H. Sonohara, M. Okuda, S. Tanaka, Y. Fujikura and S. Ito, Appl. Environ. Microbiol., 1993, 59, 1336–1341 CAS.
-
H. Takigawa, M. Okuda, J. Etsuno and Y. Fujikara, Jpn. Kokai. Tokkyo Koho, JP 06345642 A 19941220, 1994.
- P. A. García, A. B. De Oliveira and R. Batista, Molecules, 2007, 12, 455–483 CrossRef.
- R. Deng, J. Tang, L.-P. Xia, D.-D. Li, W.-J. Zhou, L.-L. Wang, G.-K. Feng, Y.-X. Zeng, Y.-H. Gao and X.-F. Zhu, Mol. Cancer Ther., 2009, 8, 873–882 CrossRef CAS PubMed.
- F. D. da Silva Araújo, L. C. de Lima Fávaro, W. L. Araújo, F. L. de Oliveira, R. Aparicio and A. J. Marsaioli, Eur. J. Org. Chem., 2012, 5225–5230 CrossRef.
- P. Ellerbrock, N. Armanino, M. K. IIg, R. Webster and D. Trauner, Nat. Chem., 2015, 7, 879–882 CrossRef CAS PubMed.
- S. P. Cook and S. J. Danishefsky, Org. Lett., 2006, 8, 5693–5695 CrossRef CAS PubMed.
- W. Dürckheimer and E. F. Paulus, Angew. Chem., Int. Ed. Engl., 1985, 24, 224–225 CrossRef.
-
(a) C. A. Carson and M. A. Kerr, Chem. Soc. Rev., 2009, 38, 3051–3060 RSC;
(b) M. Rubin, M. Marina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed.
-
(a) D. F. Taber, K. Kanai, Q. Jiang and G. Bui, J. Am. Chem. Soc., 2000, 122, 6807–6808 CrossRef CAS;
(b) S. Y. Kim, S. I. Lee, S. Y. Choi and Y. K. Chung, Angew. Chem., Int. Ed., 2008, 47, 4914–4917 CrossRef CAS PubMed.
- P. A. Wender, H. Takahashi and B. Witulski, J. Am. Chem. Soc., 1995, 117, 4720–4721 CrossRef CAS.
-
(a) B. M. Trost, F. D. Toste and H. Shen, J. Am. Chem. Soc., 2000, 122, 2379–2380 CrossRef CAS;
(b) B. M. Trost, H. C. Shen, D. B. Horne, F. D. Toste, B. G. Steinmetz and C. Koradin, Chem. – Eur. J., 2005, 11, 2577–2590 CrossRef CAS PubMed.
-
(a) G. Zuo and J. Louie, J. Am. Chem. Soc., 2005, 127, 5798–5799 CrossRef CAS PubMed;
(b) A. Fürstner, K. Majima, R. Martin, H. Krause, E. Kattnig, R. Goddard and C. W. Lehmann, J. Am. Chem. Soc., 2008, 130, 1992–2004 CrossRef PubMed;
(c) M.-C. Melcher, H. von Wachenfeldt, A. Sundin and D. Strand, Chem. – Eur. J., 2015, 21, 531–535 CrossRef CAS PubMed.
- C. Wu and N. Yoshikai, Angew. Chem., Int. Ed., 2018, 57, 6558–6562 CrossRef CAS PubMed.
-
(a) P. A. Wender, L. E. Sirois, R. T. Stemmler and T. J. Williams, Org. Lett., 2010, 12, 1604–1607 CrossRef CAS PubMed;
(b) P. A. Wender, F. Inagaki, M. Pfaffenbach and M. C. Stevens, Org. Lett., 2014, 16, 2923–2925 CrossRef CAS PubMed;
(c) P. A. Wender, C. Ebner, B. D. Fennell, F. Inagaki and B. Schroder, Org. Lett., 2017, 19, 5810–5813 CrossRef CAS PubMed.
- P. A. Wender, L. O. Haustedt, J. Lim, J. A. Love, T. J. Williams and J.-Y. Yoon, J. Am. Chem. Soc., 2006, 128, 6302–6303 CrossRef CAS PubMed.
- R. Shintani, H. Nakatsu, K. Takatsu and T. Hayashi, Chem. – Eur. J., 2009, 15, 8692–8694 CrossRef CAS PubMed.
- F. Inagaki, K. Sugikubo, Y. Miyashita and C. Mukai, Angew. Chem., Int. Ed., 2010, 49, 2206–2210 CrossRef CAS PubMed.
- P. A. Wender and D. Sperandio, J. Org. Chem., 1998, 63, 4164–4165 CrossRef CAS.
- L. Jiao, S. Ye and Z.-X. Yu, J. Am. Chem. Soc., 2008, 130, 7178–7179 CrossRef CAS PubMed.
- C.-H. Liu and Z.-X. Yu, Angew. Chem., Int. Ed., 2017, 56, 8667–8671 CrossRef CAS PubMed.
- For selected examples on the mechanistic studies of metal-catalyzed [5+2] cycloadditions, see:
(a) Z.-X. Yu, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2004, 126, 9154–9155 CrossRef CAS PubMed;
(b) Z.-X. Yu, P. H.-Y. Cheong, P. Liu, C. Y. Legault, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 2378–2379 CrossRef CAS PubMed;
(c) P. Li, P. H.-Y. Cheong, Z.-X. Yu, P. A. Wender and K. N. Houk, Angew. Chem., Int. Ed., 2008, 47, 3939–3941 CrossRef PubMed;
(d) P. Liu, L. E. Sirois, P. H.-Y. Cheong, Z.-X. Yu, I. V. Hartung, H. Rieck, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 10127–10135 CrossRef CAS PubMed;
(e) X. Xu, P. Liu, A. Lesser, L. E. Sirois, P. A. Wender and K. N. Houk, J. Am. Chem. Soc., 2012, 134, 11012–11025 CrossRef CAS PubMed;
(f) X. Hong, B. M. Trost and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 6588–6600 CrossRef CAS PubMed;
(g) X. Hong, P. Liu and K. N. Houk, J. Am. Chem. Soc., 2013, 135, 1456–1462 CrossRef CAS PubMed.
-
(a) F. W. Fowler, Angew. Chem., Int. Ed. Engl., 1971, 10, 135–136 Search PubMed;
(b) R. Herges and I. Ugi, Angew. Chem., Int. Ed. Engl., 1985, 24, 594–596 CrossRef;
(c) P.-J. Cai, F.-Q. Shi, Y. Wang, X. Li and Z.-X. Yu, Tetrahedron, 2013, 69, 7854–7860 CrossRef CAS.
-
H. Ohno, Vinylaziridines in Organic Synthesis. In Aziridines and Epoxides in Organic Synthesis, ed. A. K. Yudin, Wiley-VCH, Weinheim, 2006, pp. 37–71 Search PubMed.
- E. Kanno, K. Yamanoi, S. Koya, I. Azumaya, H. Masu, R. Yamasaki and S. Saito, J. Org. Chem., 2012, 77, 2142–2148 CrossRef CAS PubMed.
-
(a) J.-J. Feng, T.-Y. Lin, C.-Z. Zhu, H. Wang, H.-H. Wu and J. Zhang, J. Am. Chem. Soc., 2016, 138, 2178–2181 CrossRef CAS PubMed;
(b) X. Zhang, H. Zou and G. Huang, ChemCatChem, 2016, 8, 2549–2556 CrossRef CAS.
-
(a) E. E. Schultz, V. N. G. Lindsay and R. Sarpong, Angew. Chem., Int. Ed., 2014, 53, 9904–9908 CrossRef CAS PubMed;
(b) Y. Tian, Y. Wang, H. Shang, X. Xu and Y. Tang, Org. Biomol. Chem., 2015, 13, 612–619 RSC.
- J.-J. Feng, T.-Y. Lin, H.-H. Wu and J. Zhang, J. Am. Chem. Soc., 2015, 137, 3787–3790 CrossRef CAS PubMed.
- J.-J. Feng, T.-Y. Lin, H.-H. Wu and J. Zhang, Angew. Chem., Int. Ed., 2015, 54, 15854–15858 CrossRef CAS PubMed.
- M. Skvorcova, L. Grigorjeva and A. Jirgensons, Org. Lett., 2015, 17, 2902–2904 CrossRef CAS PubMed.
-
(a) B. M. Trost and S. R. Angle, J. Am. Chem. Soc., 1985, 107, 6123–6124 CrossRef CAS;
(b) J.-G. Shim and Y. Yamamoto, J. Org. Chem., 1998, 63, 3067–3071 CrossRef CAS;
(c) K. Khumtaveeporn and H. Halper, Acc. Chem. Res., 1995, 28, 414–422 CrossRef CAS;
(d) Y. D. Y. L. Getzler, V. Mahadevan, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1174–1175 CrossRef CAS PubMed;
(e) M. Miyashita, T. Mizutani, G. Tadano, Y. Iwata, M. Miyazawa and K. Tanino, Angew. Chem., Int. Ed., 2005, 44, 5094–5097 CrossRef CAS PubMed;
(f) B. M. Trost, R. C. Bunt, R. C. Lemoine and T. L. Calkins, J. Am. Chem. Soc., 2000, 122, 5968–5976 CrossRef CAS;
(g) K. Fagnou and M. Lautens, Org. Lett., 2000, 2, 2319–2321 CrossRef CAS PubMed.
- J.-J. Feng and J. Zhang, ACS Catal., 2017, 7, 1533–1542 CrossRef CAS.
- For selected examples, see:
(a) J. P. Praly, C. D. Di Stefano and L. Somsak, Tetrahedron: Asymmetry, 2000, 11, 533–537 CrossRef CAS;
(b) C. Ma, S.-J. Liu, L. Xin, J. R. Falck and D.-S. Shin, Tetrahedron, 2006, 62, 9002–9009 CrossRef CAS;
(c) W.-H. Chiou, N. Mizutani and I. Ojima, J. Org. Chem., 2007, 72, 1871–1882 CrossRef CAS PubMed;
(d) V. Sridharan, S. Maiti and J. C. Menendez, J. Org. Chem., 2009, 74, 9365–9371 CrossRef CAS PubMed;
(e) M.-F. Pouliot, L. Angers, J.-D. Hamel and J.-F. Paquin, Tetrahedron Lett., 2012, 53, 4121–4123 CrossRef CAS;
(f) M. C. Mollo and L. R. Orelli, Org. Lett., 2016, 18, 6116–6119 CrossRef CAS PubMed;
(g) W. B. Shen, X. Y. Xiao, Q. Sun, B. Zhou, X. Q. Zhu, J. Z. Yan, X. Lu and L. W. Ye, Angew. Chem., Int. Ed., 2017, 56, 605–609 CrossRef CAS PubMed.
- Y. Wu, C. Yuan, C. Wang, B. Mao, H. Jia, X. Gao, J. Lia, F. Jiang, L. Zhou, Q. Wang and H. Guo, Org. Lett., 2017, 19, 6268–6271 CrossRef CAS PubMed.
- B. N. Zhou, B. P. Ying, G. Q. Song, Z. X. Chen, J. Han and Y. F. Yan, Planta Med., 1983, 47, 35–38 CrossRef CAS PubMed.
-
(a) E. G. Li, A. M. Clark and C. D. Hufford, J. Nat. Prod., 1995, 58, 57–67 CrossRef CAS PubMed;
(b) W. C. Wang, R. F. Lu, S. X. Zhao and Z. P. Gu, Acta Pharmacol. Sin., 1988, 9, 445–448 CAS;
(c) D. J. Pan, Z. L. Li, C. Q. Hu, K. Chen, J. J. Chang and K. H. Lee, Planta Med., 1990, 56, 383–385 CrossRef CAS PubMed.
-
(a) X.-F. Gong, M.-W. Wang, S.-I. Tashiro, S. Onodera and T. Ikejima, Arch. Pharmacal Res., 2005, 28, 68–72 CrossRef CAS;
(b) J. K. S. Ko, W. C. Leung, W. K. Ho and P. Chiu, Eur. J. Pharmacol., 2007, 559, 1–3 CrossRef CAS PubMed.
-
(a) B. M. Trost, J. Waser and A. Meyer, J. Am. Chem. Soc., 2007, 129, 14556–14557 CrossRef CAS PubMed;
(b) B. M. Trost, J. Waser and A. Meyer, J. Am. Chem. Soc., 2008, 130, 16424–16434 CrossRef CAS PubMed.
- P. Ramesh, N. S. Reddy, Y. Venkateswarlue, M. V. R. Reddy and J. D. Faulkner, Tetrahedron Lett., 1998, 39, 8217–8220 CrossRef CAS.
-
J. D. Faulkner and Y. Venkateswarlue, WO0027839, 2000.
- B. M. Trost, H. M. Nguyen and C. Koradin, Tetrahedron Lett., 2011, 52, 2033–2036 CrossRef CAS.
- R. D. Witherell, K. E. O. Ylijoki and J. M. Stryker, J. Am. Chem. Soc., 2008, 130, 2176–2177 CrossRef CAS PubMed.
- K. E. O. Ylijoki, A. D. Kirk, S. Böcklein, R. D. Witherell and J. M. Stryker, Organometallics, 2015, 34, 3335–3357 CrossRef CAS.
- E. C. Garnier and L. S. Liebeskind, J. Am. Chem. Soc., 2008, 130, 7449–7458 CrossRef CAS PubMed.
- M. Harmata, Chem. Commun., 2010, 46, 8886–8903 RSC.
- M. Kudo, F. Kondo, H. Maekawa, T. Shimizu, M. Miyashita and K. Tanino, Tetrahedron Lett., 2014, 55, 1192–1195 CrossRef CAS.
- K. Mitachi, T. Yamamoto, F. Kondo, T. Shimizu, M. Miyashita and K. Tanino, Chem. Lett., 2010, 39, 630–632 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Yong-Gang
Zhang
a,
Zhiming
Wang
a,
Yong-Gang
Zhang
a,
Zhiming
Wang