
Laser synthesis of oxygen vacancy-modified CoOOH for highly efficient oxygen evolution†
Chao
Meng
a,
Mengchang
Lin
a,
Xuechun
Sun
b,
Xiaodong
Chen
a,
Xuemin
Chen
c,
Xiwen
Du
 *b and
Yue
Zhou
*a
*b and
Yue
Zhou
*a
aCollege of Electrical Engineering and Automation, Shandong University of Science and Technology, Qingdao, 266590, China
bInstitute of New-Energy Materials, School of Materials Science and Engineering, Tianjin University, Tianjin, 300072, China. E-mail: xwdu@tju.edu.cn
cCollege of Science, Hebei University of Science & Technology, Shijiazhuang, 050018, China
First published on 16th January 2019
Abstract
Introducing oxygen vacancies into transition-metal oxide materials would improve their catalytic activity but usually needs high-temperature or high-pressure conditions, and multi-step procedures, and thus are time consuming and not energy efficient. Herein, laser ablation in liquids (LAL), a green, mild and effective approach, has been, for the first time, employed to prepare CoOOH nanosheets with abundant oxygen vacancies and relatively thin thickness. Our theoretical and experimental results demonstrate that oxygen vacancies can optimize the absorption of oxygen evolution reaction (OER) intermediates and improve electrical conductivity; meanwhile, the relatively thin thickness can provide more active sites, thus leading to excellent OER activity of oxygen vacancy-modified CoOOH nanosheets. This work may provide guidance for exploring other efficient non-noble metal catalysts for water oxidation.
The OER is the bottleneck in electrochemical water splitting and metal–air batteries, due to its sluggish kinetics and requirement of large overpotentials.1,2 For this reason, numerous efforts have been made in developing high-performance catalysts to promote the OER process. It is known that noble metal oxides like IrO2 and RuO2 are the state-of-the-art electrocatalysts for the OER, but their high cost and poor durability limit their large-scale applications.3,4 Recently, transition-metal (oxy)hydroxides (MOOH; M = Co, Ni, Fe, Mn, etc.) have been accepted as a class of promising OER catalysts in alkaline media, thanks to their large reserves and excellent electrochemical stability.5,6 Unfortunately, the catalytic efficiency of pristine MOOH is still below what is expected, with a higher overpotential and slower reaction kinetics than noble metal oxide catalysts.
Both theoretical calculations and experimental results demonstrate that the introduction of oxygen vacancies is an effective strategy to improve the OER catalytic efficiencies of metal oxides, hydroxides, and MOOH.7,8 The oxygen vacancies can result in an optimized coordination environment of metal sites, which improves the electrical conductivity and the absorption of OER intermediates, thus accelerating the OER.9,10 So far, approaches including hydrogen annealing,11,12 plasma13,14 and flame treatments15,16 have shown their efficiency in creating oxygen vacancies. However, these approaches usually involve high-temperature or high-pressure processes, and thus are not energy efficient and easily cause excessive destruction of material structure.17,18 Recently, a mild synthetic method was developed to form oxygen vacancies by immersing the sample in NaBH4 solution.19 Although enough oxygen vacancies have been induced to largely enhance the performance of the catalyst, this method still needs the multi-step procedure.15 Therefore, it is highly desired to find a green and mild one-step approach to introducing abundant oxygen vacancies.
LAL is known as a green, simple and rapid technique for the synthesis of materials under nominally ambient conditions.20 Intriguingly, LAL can generate extremely high temperature and pressure at the moment of laser ablation, followed by a rapid cooling process after laser irradiation, which benefit the formation and stability of defects.21,22 Our previous work reported the capability of LAL to create plentiful oxygen vacancies into cobalt oxides.23 In this work, we first employ LAL to synthesize oxygen vacancy-modified CoOOH nanosheets by irradiating the Co target immersed in KOH solution. Our theoretical and experimental results demonstrate that abundant oxygen vacancies can obviously improve the absorption of OER intermediates and electrical conductivity. Meanwhile, the relatively thin thickness can provide more active sites, thus synthetically leading to an excellent OER performance of the catalyst. Resultantly, the oxygen vacancy-modified CoOOH nanosheets show a low overpotential of 330 mV at a current density of 10 mA cm−2, a small Tafel slope of 63.2 mV dec−1, and high catalytic durability, which all exceed those of commercial RuO2. This work comes up with the possibilities of popularizing LAL technology in designing MOOH with plentiful oxygen vacancies as efficient catalysts.
The synthetic procedure is schematically illustrated in Fig. 1a–c. Firstly, the Co target as the raw material was placed in 1 M KOH solution, and then irradiated using a nanosecond laser for 20 minutes to obtain the so-called L-CoOOH sample, which has a yield of about 5.7 mg h−1 (Fig. S1, ESI†). Finally, L-CoOOH was utilized for catalyzing the OER (see details in the Experimental section of the ESI†). X-ray diffraction (XRD) was adopted to analyze the phase and composition of L-CoOOH. As shown in Fig. 1d, the main peaks at 20.239°, 38.887° and 50.583° are perfectly indexed to the (003), (012) and (015) planes of rhombohedral phase CoOOH (JCPDS no. 07-0169), respectively, indicating the successful synthesis of crystalline CoOOH, which is further confirmed by the energy-dispersive spectroscopy (EDS) and X-ray photoelectron spectroscopy (XPS) spectra in Fig. S2 (ESI†). Furthermore, the morphology of L-CoOOH was examined by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) (Fig. S3, ESI†). The low-magnification TEM image (Fig. 1e) illustrates that the L-CoOOH sample possesses a hexagonal sheet-like nanostructure with a width range of 0.3–1.0 μm. The corresponding selected-area electron diffraction (SAED) pattern (inset in Fig. 1f) clearly exhibits a set of diffraction spots, revealing the highly crystalline nature of L-CoOOH. In order to learn more about the structure of L-CoOOH, an individual L-CoOOH nanosheet was characterized by high-resolution TEM (HRTEM, Fig. 1f). The result shows that a clearly identified lattice fringe space of 0.23 nm corresponds to the (012) plane of rhombohedral CoOOH, agreeing with the XRD analysis. Meanwhile, some subtle lattice distortions are observed in L-CoOOH, which may benefit OER catalysis.24,25
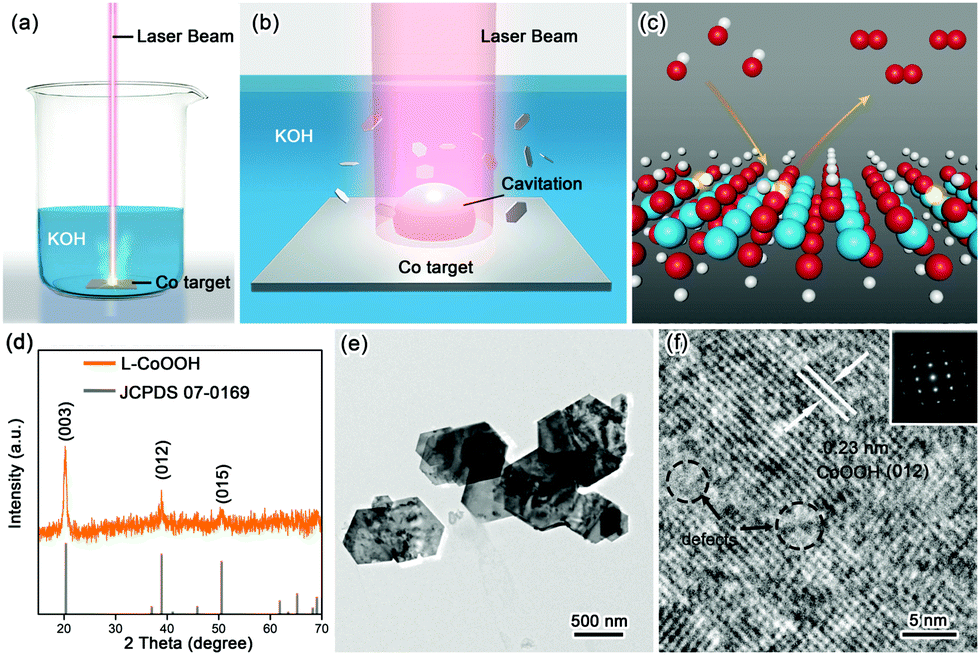 | ||
| Fig. 1 Synthesis and characterization of L-CoOOH. (a) Schematic illustration of the synthesis of L-CoOOH via LAL and (b) the corresponding magnification of the laser irradiation region. (c) Schematic representation of L-CoOOH with abundant oxygen vacancies for the OER. Color code: white for H, red for O, cyan for Co, and transparent color for vacancy. (d) XRD pattern of L-CoOOH. (e and f) Low-magnification TEM and HRTEM images of L-CoOOH. The inset in (f) shows the SAED pattern of L-CoOOH. | ||
The OER performance of L-CoOOH was evaluated using a typical three-electrode system in 1 M KOH (Fig. S5, ESI†), and bulk CoOOH (B-CoOOH) and commercial RuO2 were used as the references (Fig. S4, ESI†). All the linear sweep voltammetry (LSV) curves were obtained at the scan rate of 5 mV s−1. As shown in Fig. 2a, B-CoOOH is active for the OER with the need for an overpotential of 410 mV to drive 10 mA cm−2. In sharp contrast, L-CoOOH exhibits much higher activity, only requiring an overpotential of 330 mV to reach 10 mA cm−2, and even outperforms RuO2 (340 mV). Furthermore, the Tafel slope is considered as a crucial indicator to evaluate the kinetic information for the OER. According to the Tafel equation (η = b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logj + a, where η is the overpotential, b is the Tafel slope, j is the current density and a is the constant),26 the Tafel slope of L-CoOOH is 63.2 mV dec−1, which is much lower than that of B-CoOOH (75.4 mV dec−1) and even close to that of RuO2 (64.9 mV dec−1), suggesting favourable OER kinetics for L-CoOOH. Eventually, considering the overpotential at 10 mA cm−2 and the Tafel slope comprehensively, the OER performance of L-CoOOH is better than or comparable with those of most previously reported Co-based OER catalysts in alkaline solutions (Fig. 2c and Table S1, ESI†).
logj + a, where η is the overpotential, b is the Tafel slope, j is the current density and a is the constant),26 the Tafel slope of L-CoOOH is 63.2 mV dec−1, which is much lower than that of B-CoOOH (75.4 mV dec−1) and even close to that of RuO2 (64.9 mV dec−1), suggesting favourable OER kinetics for L-CoOOH. Eventually, considering the overpotential at 10 mA cm−2 and the Tafel slope comprehensively, the OER performance of L-CoOOH is better than or comparable with those of most previously reported Co-based OER catalysts in alkaline solutions (Fig. 2c and Table S1, ESI†).
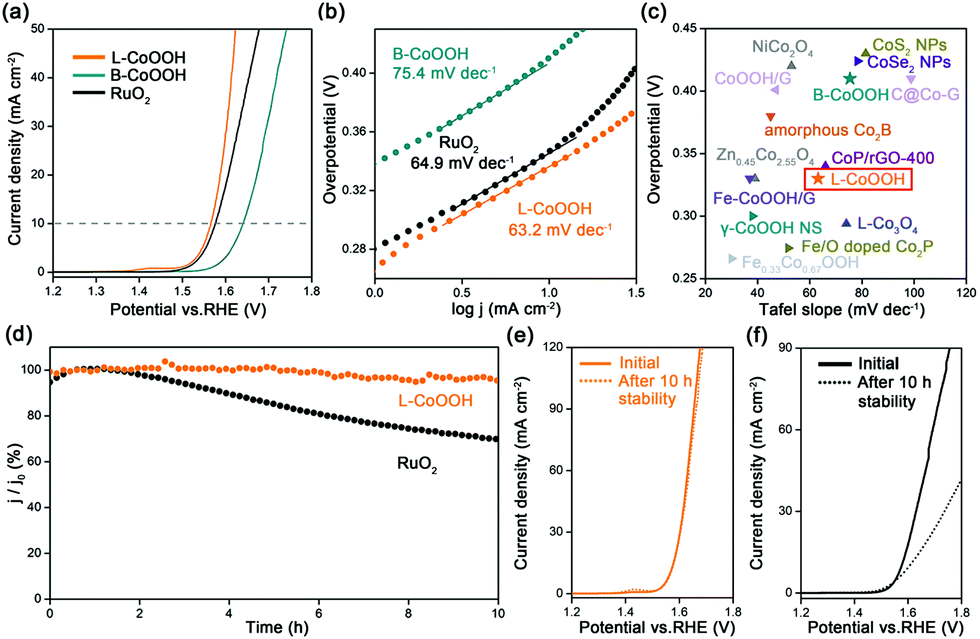 | ||
| Fig. 2 Electrocatalytic OER performance of L-CoOOH. (a) LSV curves of L-CoOOH, B-CoOOH and RuO2 recorded in 1 M KOH. (b) Tafel plots obtained from the LSV curves in (a). (c) Comparison of Tafel slopes and overpotentials at 10 mA cm−2 for various Co-based catalysts reported in the literature, B-CoOOH and L-CoOOH in 1 M KOH. (d) OER chronoamperometric responses of L-CoOOH and RuO2 at overpotentials of 330 mV and 340 mV, respectively. LSV curves of (e) L-CoOOH and (f) RuO2 before and after the stability test. | ||
In addition, long-term durability is also a key criterion for energy conversion application. In this work, the long-term durability of the catalyst was investigated by chronoamperometric measurement. After a 10 h test, RuO2 only maintains 69% of its initial current density (Fig. 2d), and the LSV plot is far away from the pristine one (Fig. 2f). In comparison, L-CoOOH maintains a high retention ratio of 95% of its initial current density (Fig. 2d), and the LSV plot is very close to the original one (Fig. 2e), revealing the superior durability of L-CoOOH over RuO2. Moreover, the nearly unchanged morphology and composition of L-CoOOH after the OER durability test are shown in Fig. S6 (ESI†), further demonstrating the stable structure of L-CoOOH.
To discover the origin of the excellent performance of L-CoOOH, we first measured the electrochemical double-layer capacitance (Cdl) on two CoOOH catalysts, which is determined by the slope of the linear relationship between the current density and the scan rate. As shown in Fig. 3a and Fig. S7 (ESI†), the Cdl value of L-CoOOH (10.24 mF cm−2) is 7.4-fold higher than that of B-CoOOH (1.38 mF cm−2), implying the existence of more active sites in L-CoOOH.17 Besides, the electrical conductivity of the two CoOOH catalysts was measured by electrochemical impendence spectroscopy (EIS). The semi-circular diameter of L-CoOOH is much smaller than that of B-CoOOH, due to the smaller charge transfer resistance (Rct) in L-CoOOH (Fig. 3b). By fitting EIS plots using the equivalent circuit in the inset of Fig. 3b, the Rct values of B-CoOOH and L-CoOOH can be determined as 97.2 Ω and 47.6 Ω, respectively (Table S2, ESI†), suggesting a faster electron transfer in L-CoOOH during OER catalysis.27
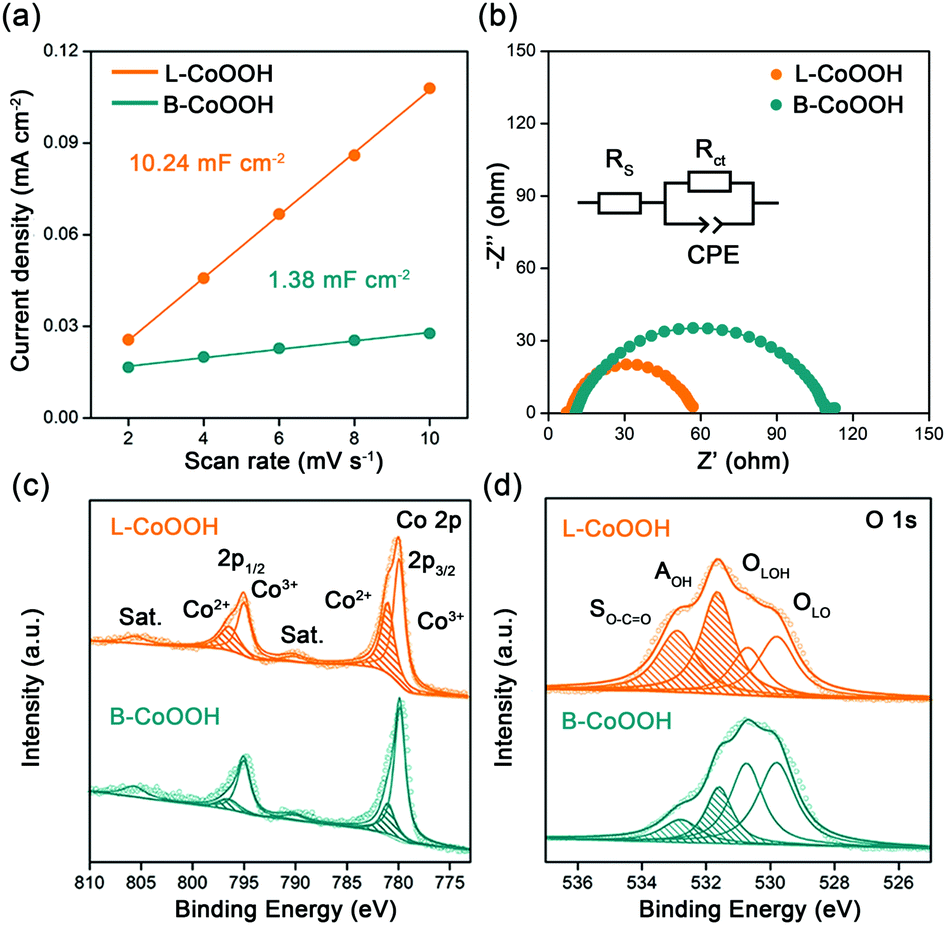 | ||
| Fig. 3 Origin of the excellent OER activity of L-CoOOH. (a) The relationship between the current density and the scan rate. (b) The EIS of L-CoOOH and B-CoOOH recorded at a potential of 1.56 VRHE. (c) Co 2p and (d) O 1s XPS spectra of L-CoOOH and B-CoOOH. | ||
To find the intrinsic reason behind the increased active sites and electrical conductivity of L-CoOOH, X-ray photoelectron spectroscopy (XPS) was first employed to explore the surface valence states of the two CoOOH catalysts. In the high-resolution Co 2p spectra (Fig. 3c), the Co 2p3/2 and Co 2p1/2 peaks are deconvoluted into Co2+ and Co3+. The peaks at 779.8 and 795 eV correspond to Co3+, while those at 781 and 796.5 eV correspond to Co2+, which agree with previous reports.18,28 Obviously, the Co2+ peaks of L-CoOOH are higher than those of B-CoOOH, indicating more oxygen vacancies in L-CoOOH, which provides more naked Co3+ as active sites for the OER.29 Besides, the O 1s spectra were split into four peaks at 529.8, 530.7, 531.7 and 532.7 eV, corresponding to lattice O (OLO), lattice OH (OLOH), absorbed OH (AOH), and surface O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (SO–CO), respectively (Fig. 3d).30 As listed in Table S3 (ESI†), the relative ratios of OLO and OLOH in L-CoOOH are both lower than those in B-CoOOH, confirming more oxygen vacancies in L-CoOOH. Electron paramagnetic resonance (EPR) spectroscopy was also performed to consolidate the above results. Fig. S8 (ESI†) shows the presence of a symmetrical EPR signal (g = 2.003) in CoOOH, which originates from the unpaired electrons at oxygen vacancy sites.31 More notably, the EPR signal intensity of L-CoOOH is significantly higher than that of B-CoOOH, further confirming the existence of more oxygen vacancies in L-CoOOH.32,33
O (SO–CO), respectively (Fig. 3d).30 As listed in Table S3 (ESI†), the relative ratios of OLO and OLOH in L-CoOOH are both lower than those in B-CoOOH, confirming more oxygen vacancies in L-CoOOH. Electron paramagnetic resonance (EPR) spectroscopy was also performed to consolidate the above results. Fig. S8 (ESI†) shows the presence of a symmetrical EPR signal (g = 2.003) in CoOOH, which originates from the unpaired electrons at oxygen vacancy sites.31 More notably, the EPR signal intensity of L-CoOOH is significantly higher than that of B-CoOOH, further confirming the existence of more oxygen vacancies in L-CoOOH.32,33
Then we analyze the reasons why oxygen vacancies can be created in L-CoOOH by LAL, which are mainly attributed to the following two aspects. On the one hand, when a laser beam with energy higher than the ablation threshold irradiates the Co target in the solution, their interaction will immediately produce a Co-enriched plasma with high temperature and high pressure.20,34 As predicted by Hagemark's defect reaction constants, if the vacancies are formed in equilibrium states at every instant time with fixed temperature, the equilibrium concentration of vacancies will exponentially increase with temperature.35,36 Therefore, the high temperature, high pressure and Co-enriched plasma is favourable for forming oxygen vacancies in L-CoOOH. On the other hand, the formation of L-CoOOH nanosheets is very fast due to the huge temperature difference between the hot plasma and the surrounding solution.37,38 According to recent research results, the higher the formation rate of nanomaterials, the more defects they have.38,39 Thus, the high concentration of oxygen vacancies formed at high temperature is quickly “frozen” in the CoOOH lattice.
However, it is worth noting that L-CoOOH (∼12 nm) is relatively thinner than B-CoOOH (∼65 nm) in Fig. S9 (ESI†), which may also contribute to more active sites and higher electrical conductivity in L-CoOOH. In order to verify the above possibility, Reference CoOOH (R-CoOOH) was introduced as a reference with the same size as L-CoOOH (Fig. S10, ESI†) and similar oxygen vacancy concentration to B-CoOOH (Fig. S8 and S11 and Table S3, ESI†). As can be seen in Fig. S12 and Table S2 (ESI†), the overpotential at 10 mA cm−2 of R-CoOOH (379 mV) slightly surpasses that of B-CoOOH by 31 mV, mainly owing to the higher Cdl value of R-CoOOH (6.46 mF cm−2). These collective results reveal that the thinner thickness only leads to more active sites, but not to better electrical conductivity of L-CoOOH.
Besides experimental investigations, a series of density functional theory (DFT) calculations were also performed to get a fundamental understanding of the correlation between the oxygen vacancies of CoOOH and OER performance. Only considering the four-electron (4e) reaction pathway as shown in eqn (S1)–(S4) in the ESI,† we first employed the computational hydrogen electrode model to compute the Gibbs free energy of three OER intermediates (*OH, *O, and *OOH) for CoOOH with a perfect surface and CoOOH with oxygen vacancies (see Computational details in the ESI†). The corresponding two CoOOH structures are shown in Fig. 4a and b. According to computational results in Fig. S13 (ESI†) and Fig. 4c, the largest Gibbs free energy difference (Step III) is the rate determining step, which is the same for CoOOH with a perfect surface and CoOOH with oxygen vacancies. The Gibbs free energy difference value of CoOOH with oxygen vacancies (ΔGIII = ΔG*OOH − ΔG*O = 1.62 eV) is smaller than that of CoOOH with a perfect surface (ΔGIII = 2.15 eV). In other words, CoOOH with oxygen vacancies requires a lower overpotential to drive water oxidation. Therefore, our computational results indicate that the OER catalytic activity of CoOOH can be pronouncedly improved by introducing oxygen vacancies, which are consistent with experimental results. Moreover, the electronic band structures of CoOOH without and with oxygen vacancies were analyzed in terms of projected densities of states (PDOS). As indicated in Fig. S14 (ESI†) and Fig. 4d, when oxygen vacancies are introduced in CoOOH, some new electronic states appear near the Fermi level, leading to higher electrical conductivity of CoOOH.40
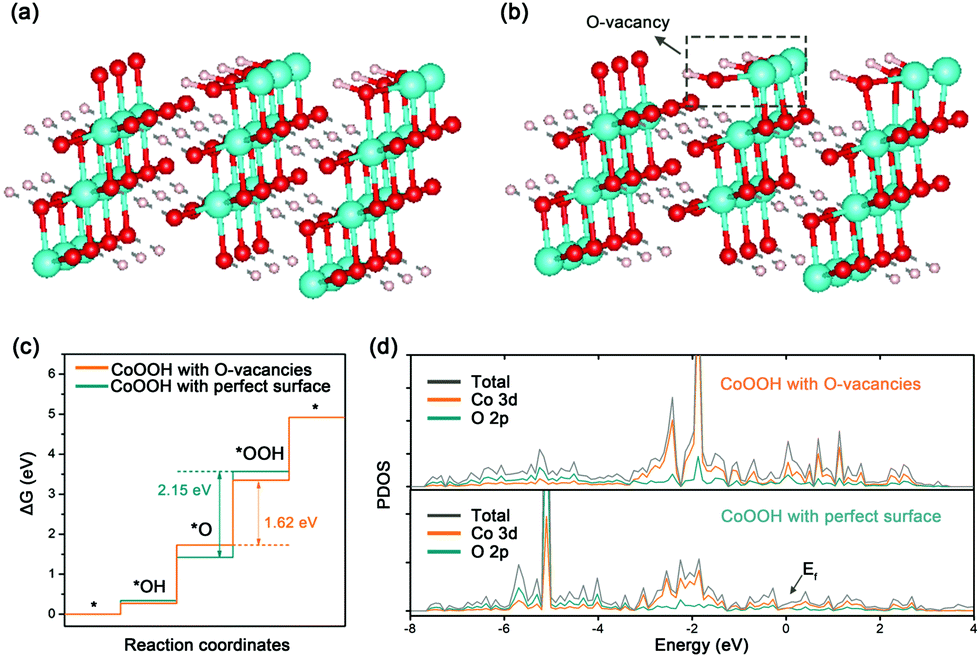 | ||
| Fig. 4 DFT calculations of CoOOH with a perfect surface and CoOOH with oxygen vacancies. The structure models of (a) a perfect CoOOH (012) surface and (b) a CoOOH (012) surface with oxygen vacancies. Color code: white for H, red for O, and cyan for Co. (c) The free energy profile for the OER pathway and (d) PDOS of the perfect surface and the surface with oxygen vacancies. | ||
In view of the above, the excellent OER performance of L-CoOOH can be attributed to three aspects. First, oxygen vacancies on the surface of L-CoOOH optimize the absorption of OER intermediates, which, in turn, accelerates the whole OER process. Second, the electrical conductivity of L-CoOOH is also enhanced by oxygen vacancies, thus ensuring highly efficient electron transport. Last but not the least, the relatively thin thickness of L-CoOOH provides more active sites, which benefits OER catalysis.
In summary, we have, for the first time, employed LAL to introduce abundant oxygen vacancies into CoOOH nanosheets, and identified their role in OER catalysis from both theoretical and experimental points of view. We have demonstrated that the laser induced oxygen vacancies can largely improve the absorption of OER intermediates and enhance their electrical conductivity. Meanwhile, their relatively thin thickness can provide more active sites; resultantly, the L-CoOOH nanosheets exhibit excellent OER activity, outperforming commercial RuO2. Beyond the synthesis of oxygen vacancy-modified CoOOH nanosheets, LAL may be a green and mild one-step approach for producing other MOOH with plentiful oxygen vacancies.
This work was supported by the Scientific Research Foundation of Shandong University of Science and Technology for Recruited Talents (2017RCJJ057), the Natural Science Foundation of Hebei Province (No. B2018208090) and the Natural Science Foundation of China (51671141, 51571149, 51471115, and 21573117).
Conflicts of interest
The authors declare no competing financial interest.References
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- D. McAteer, I. J. Godwin, Z. Ling, A. Harvey, L. He, C. S. Boland, V. Vega-Mayoral, B. Szydłowska, A. A. Rovetta, C. Backes, J. B. Boland, X. Chen, M. E. G. Lyons and J. N. Coleman, Adv. Energy Mater., 2018, 8, 1702965 CrossRef.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CAS.
- D. Y. Kuo, J. K. Kawasaki, J. N. Nelson, J. Kloppenburg, G. Hautier, K. M. Shen, D. G. Schlom and J. Suntivich, J. Am. Chem. Soc., 2017, 139, 3473–3479 CrossRef CAS PubMed.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CAS.
- F. Li, J. Du, X. Li, J. Shen, Y. Wang, Y. Zhu and L. Sun, Adv. Energy. Mater., 2018, 8, 1702598 CrossRef.
- M. Q. Yang, J. Wang, H. Wu and G. W. Ho, Small, 2018, 14, e1703323 CrossRef PubMed.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- F. Song, K. Schenk and X. Hu, Energy Environ. Sci., 2016, 9, 473–477 RSC.
- J. Liu, J. Nai, T. You, P. An, J. Zhang, G. Ma, X. Niu, C. Liang, S. Yang and L. Guo, Small, 2018, 14, e1703514 CrossRef PubMed.
- Y. Ou, W. Tian, L. Liu, Y. Zhang and P. Xiao, J. Mater. Chem. A, 2018, 6, 5217–5228 RSC.
- T. Pussacq, H. Kabbour, S. Colis, H. Vezin, S. Saitzek, O. Gardoll, C. Tassel, H. Kageyama, C. Laberty Robert and O. Mentré, Chem. Mater., 2017, 29, 1047–1057 CrossRef CAS.
- L. Rong, W. Yanyong, L. Dongdong, Z. Yuqin and W. Shuangyin, Adv. Mater., 2017, 29, 1701546 CrossRef PubMed.
- X. Lei, J. Qianqian, X. Zhaohui, L. Xingyue, H. Jia, W. Shuangyin and D. Liming, Angew. Chem., 2016, 128, 5363–5367 CrossRef.
- D. Zhou, X. Xiong, Z. Cai, N. Han, Y. Jia, Q. Xie, X. Duan, T. Xie, X. Zheng, X. Sun and X. Duan, Small Methods, 2018, 2, 1800083 CrossRef.
- I. S. Cho, M. Logar, C. H. Lee, L. Cai, F. B. Prinz and X. Zheng, Nano Lett., 2014, 14, 24–31 CrossRef CAS PubMed.
- Z. Cai, Y. Bi, E. Hu, W. Liu, N. Dwarica, Y. Tian, X. Li, Y. Kuang, Y. Li, X. Q. Yang, H. Wang and X. Sun, Adv. Energy. Mater., 2018, 8, 1701694 CrossRef.
- W. Xu, F. Lyu, Y. Bai, A. Gao, J. Feng, Z. Cai and Y. Yin, Nano Energy, 2018, 43, 110–116 CrossRef CAS.
- Z. Linzhou, G. Lei, Y. Yisu, L. Mengran, J. Yi, Y. Xiangdong and Z. Zhonghua, Adv. Mater., 2017, 29, 1606793 CrossRef PubMed.
- H. Zeng, X. W. Du, S. C. Singh, S. A. Kulinich, S. Yang, J. He and W. Cai, Adv. Funct. Mater., 2012, 22, 1333–1353 CrossRef CAS.
- H. Palneedi, J. H. Park, D. Maurya, M. Peddigari, G. T. Hwang, V. Annapureddy, J. W. Kim, J. J. Choi, B. D. Hahn, S. Priya, K. J. Lee and J. Ryu, Adv. Mater., 2018, 30, e1705148 CrossRef PubMed.
- C. Dong, Z. W. Liu, J. Y. Liu, W. C. Wang, L. Cui, R. C. Luo, H. L. Guo, X. L. Zheng, S. Z. Qiao, X. W. Du and J. Yang, Small, 2017, 13, 1603903 CrossRef PubMed.
- Y. Zhou, C. K. Dong, L. L. Han, J. Yang and X. W. Du, ACS Catal., 2016, 6, 6699–6703 CrossRef CAS.
- Y. Sun, K. Xu, Z. Wei, H. Li, T. Zhang, X. Li, W. Cai, J. Ma, H. J. Fan and Y. Li, Adv. Mater., 2018, 30, 1802121 CrossRef PubMed.
- L. An, Y. Li, M. Luo, J. Yin, Y. Q. Zhao, C. Xu, F. Cheng, Y. Yang, P. Xi and S. Guo, Adv. Funct. Mater., 2017, 27, 1703779 CrossRef.
- J. Nai, Y. Lu, L. Yu, X. Wang and X. W. Lou, Adv. Mater., 2017, 29, 1703870 CrossRef PubMed.
- C. Meng, T. Ling, T. Y. Ma, H. Wang, Z. Hu, Y. Zhou, J. Mao, X. W. Du, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2017, 29, 1604607 CrossRef PubMed.
- L. Yang, H. Zhou, X. Qin, X. Guo, G. Cui, A. M. Asiri and X. Sun, Chem. Commun., 2018, 54, 2150–2153 RSC.
- Y. Li, F. M. Li, X. Y. Meng, S. N. Li, J. H. Zeng and Y. Chen, ACS Catal., 2018, 8, 1913–1920 CrossRef CAS.
- J. Wang, J. Liu, B. Zhang, H. Wan, Z. Li, X. Ji, K. Xu, C. Chen, D. Zha, L. Miao and J. Jiang, Nano Energy, 2017, 42, 98–105 CrossRef CAS.
- F. Tang, W. Cheng, Y. Huang, H. Su, T. Yao, Q. Liu, J. Liu, F. Hu, Y. Jiang, Z. Sun and S. Wei, ACS Appl. Mater. Interfaces, 2017, 9, 26867–26873 CrossRef CAS PubMed.
- Y. C. Zhang, Z. Li, L. Zhang, L. Pan, X. Zhang, L. Wang, A. Fazale and J. J. Zou, Appl. Catal., B, 2018, 224, 101–108 CrossRef CAS.
- Y. Wang, J. Cai, M. Wu, J. Chen, W. Zhao, Y. Tian, T. Ding, J. Zhang, Z. Jiang and X. Li, Appl. Catal., B, 2018, 239, 398–407 CrossRef CAS.
- M. Dell’Aglio, R. Gaudiuso, O. De Pascale and A. De Giacomo, Appl. Surf. Sci., 2015, 348, 4–9 CrossRef.
- K. I. Hagemark and L. C. Chacka, J. Solid State Chem., 1975, 15, 261–270 CrossRef CAS.
- K. I. Hagemark, J. Solid State Chem., 1976, 16, 293–299 CrossRef CAS.
- M. Rui, X. Li, L. Gan, T. Zhai and H. Zeng, Adv. Funct. Mater., 2016, 26, 5051–5060 CrossRef CAS.
- D. Zhang, J. Liu, P. Li, Z. Tian and C. Liang, ChemNanoMat, 2017, 3, 512–533 CrossRef CAS.
- D. Zhang, B. Gökce and S. Barcikowski, Chem. Rev., 2017, 117, 3990–4103 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang, Z. Liu, C. Xie, S. Feng, D. Liu, M. Shao and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 5867–5871 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc08951e |
| This journal is © The Royal Society of Chemistry 2019 |