
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControl of helical chirality in supramolecular chromophore–DNA architectures†
Robert
Hofsäβ
,
Philipp
Ensslen
and
Hans-Achim
Wagenknecht
 *
*
Institute of Organic Chemistry, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 6, 76131 Karlsruhe, Germany. E-mail: Wagenknecht@kit.edu
First published on 8th January 2019
Abstract
Four different D- and L-configured chromophore–2′-deoxyuridine conjugates were applied to elucidate the helical chirality of their non-covalent assemblies along the D- and L-configured DNA templates by optical spectroscopy. There is no configuration-selective recognition between these nucleosides and the DNA templates. The helicity of the DNA assemblies is either controlled by the configuration of the DNA template or by the nucleoside configuration.
The regular right-handed helical structure of DNA is spontaneously formed by two complementary oligonucleotides and shows well-defined π–π stacking distances between the base pairs and sequence-selective hydrogen bonding encoded by the canonical pairing rules. These two major interactions drive also the formation of the majority of known DNA nanoarchitectures, as pioneered by Seeman and Rothemund,1,2 in a programmable way.3–5 The covalent attachment of chromophores to the nucleotides as DNA building blocks adds promising light-harvesting or optoelectronic properties to DNA by structurally designed photophysics.6–9 In order to build new and defined chromophore–DNA architectures supramolecular oligomerization of building blocks is the straightforward bottom-up approach.10 It is synthetically less challenging because it avoids the “bottleneck” of solid-phase oligonucleotide synthesis and purification for covalent connections between the building blocks.11 For this supramolecular approach, the Watson–Crick base pairing of single-stranded DNA templates together with chromophore-enhanced π–π stacking drives the self-assembly of chromophores. By these means, Schenning et al.12–14 and Balaz et al.15,16 assembled naphthalenes, oligo-p-phenylenevinylenes, or porphyrins non-covalently along single-stranded DNA templates. Häner et al. even prepared functional DNA-grafted polymers.17,18 We evidenced the assembly of ethnyl pyrenes19 and ethynyl nile reds20 as 2′-deoxyuridine conjugates specifically along oligo-2′-deoxyadenosines as DNA templates to obtain supramolecular DNA–chromophore assemblies as functional photoactive layers in solar cells.21 Most recently, we showed that ethynyl pyrene and ethynyl nile red can be assembled in a sequence-programmable fashion controlled by the DNA template.22
The construction of π-functional supramolecular chromophore assemblies faces the challenge that chromophores have to be kept in close proximity but complete self-quenching has to be ruled out. Helical stacking is an important solution to this problem, and therefore, the helicity is the most important key feature of double-stranded DNA for chromophore architectures, as shown, for instance, with triphenylamines.23 The helical twist controls the rate and efficiency of energy and electron transfer processes and reduces the complete self-quenching that is typically observed in non-helical aggregates. This principle guided us, for instance, to develop a white-light emitting DNA18,24 and to use such DNA architectures in solar cells.21 The key and basic question is how helical chirality is controlled in such supramolecular DNA–chromophore architectures. In principle, chirality may be governed by the 2′-desoxyribofuranoside configuration of the DNA template, which is either D or L. It is known that both unmodified DNA double helices possess the same conformation and dynamic properties except for chirality. The higher order structures of double-stranded L-DNA are also the exact mirror images (left-handed helices) of that of natural D-DNA.25,26 To our knowledge, however, L-configured chromophore–nucleoside conjugates were not studied before for supramolecular assemblies. Herein, we follow this path and present a new study to elucidate the origin of chirality in DNA-templated chromophore assemblies. We used a combination of two different D- and L-configured chromophor–nucleoside conjugates, 1D/1L and 2D/2L, as building blocks which are non-covalently assembled along D- and L-configured DNA templates to study the chirality of supramolecular DNA architectures by means of optical spectroscopy.
The four chromophore–nucleosides differ by the attached chromophore, either ethynylpyrene (1D and 1L) or ethynyl nile red (2D and 2L), and by the configuration of the 2′-deoxyribofuranosides, either D or L (Fig. 1). The D-configured conjugates 1D and 2D were described previously;19,20 the L-configured conjugates were synthesized accordingly (see ESI† and Fig. S2 and S3). Stock solutions in DMSO were prepared for each of the four nucleosides. The absorbance and fluorescence properties of the new L-configured nucleosides were nearly identical (within the experimental error) to those of the D-configured ones (see Fig. S16–S19, ESI†). The DNA-templated self-assemblies were prepared according to the following procedure (Fig. 1). Small amounts of the nucleoside stock solutions in DMSO were added to the aqueous solution containing the DNA templates, which are the D-configured A20 or the L-configured a20. Not more than 2% DMSO were added to the final DNA samples in water. Notably, all four nucleoside conjugates were nearly completely insoluble in water, and only those nucleosides that were bound by specific base pairing to the given DNA template were kept in aqueous solution. We know from previous studies that 1D and 2D specifically bind only to oligo-2′-deoxyadenosines as correct templates and not to oligothymidines as wrong templates.20 This represents an important advantage for the preparation of our supramolecular chromophore assemblies along DNA templates, because excess, hence unbound chromophores could simply be removed from the sample by short centrifugation (1 min at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, Fig. 1). A DNA-typical annealing procedure as previously described19 is not necessary to form DNA assemblies with nearly complete occupation of binding sites.20 This indicates a thermodynamically driven assembly. After centrifugation, the supernatants of the samples were studied by means of optical spectroscopy.
000g, Fig. 1). A DNA-typical annealing procedure as previously described19 is not necessary to form DNA assemblies with nearly complete occupation of binding sites.20 This indicates a thermodynamically driven assembly. After centrifugation, the supernatants of the samples were studied by means of optical spectroscopy.
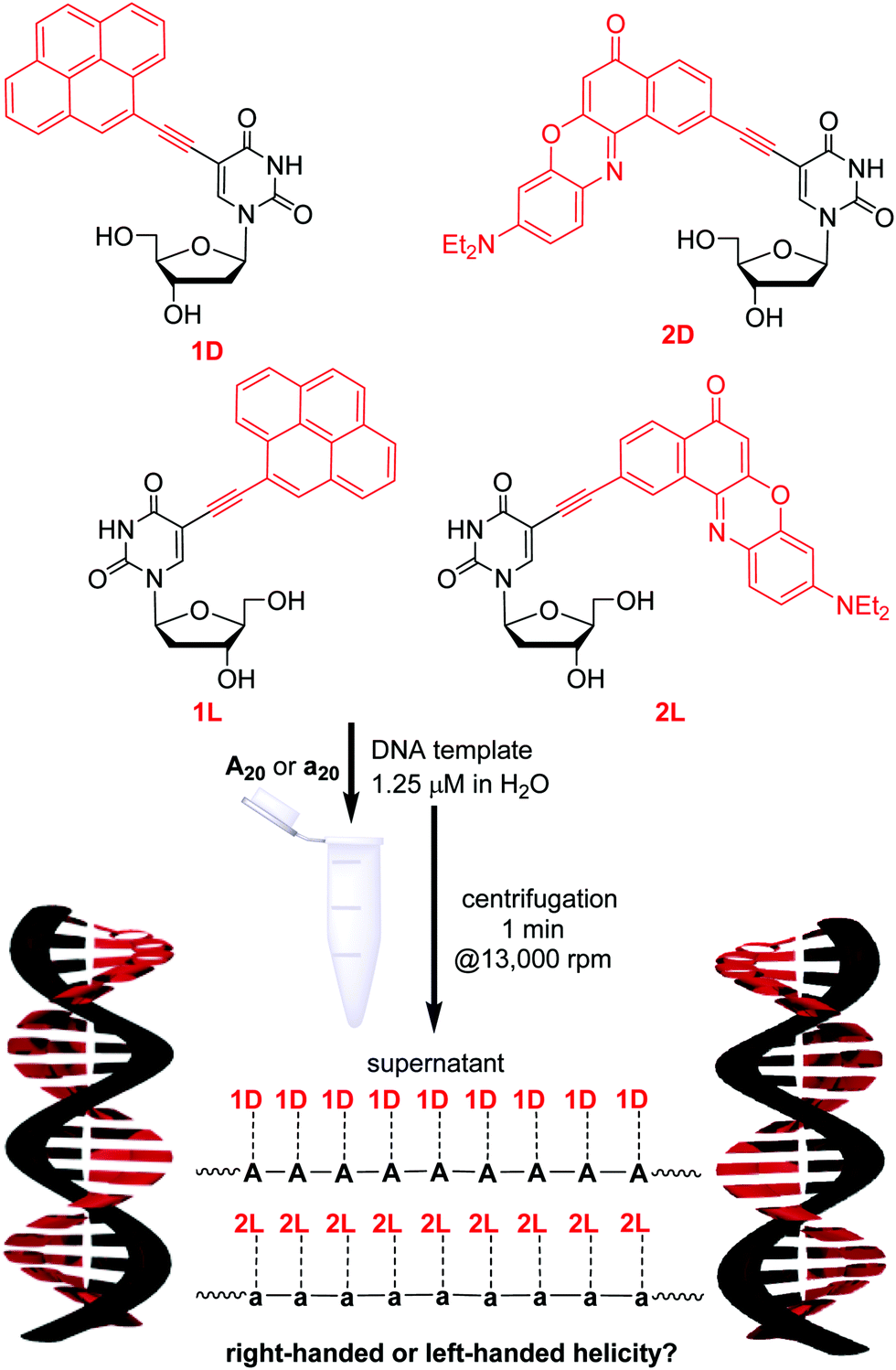 | ||
| Fig. 1 Structure of 1D, 2D, 1L and 2L, and preparation of chromophore assemblies along single-stranded DNA templates A20 (with D-configuration) and a20 (with L-configuration), and idealized schematic drawing of the assemblies of 1D along A20 and of 2L along a20. | ||
Firstly, we checked if the nucleosides show selective binding to the differently configured DNA templates. The DNA templates A20 and a20 are complementary to all four chromophore–2′-deoxyuridine conjugates. Titration experiments were carried out to determine how many binding sites at these DNA templates are occupied. Each chromophore–nucleoside aliquot was equal to the binding of one nucleoside to the template (see Fig. S22 and S23, ESI†). Expectedly, the absorption of the supernatant indicated high occupancy rates for the assemblies with matching configurations between nucleosides and DNA templates (Table 1): 90% of binding sites on A20 are occupied by 1D and 80% of binding sites on a20 are occupied by 1L. 2D and 2L bind even quantitatively to their configuration-matching templates A20 and a20, respectively, indicating a stronger π–π-stacking interaction between ethynyl nile red chromophores compared to ethynylpyrenes. Surprisingly, high occupancy rates were also observed for those DNA assemblies in which the configurations do not match between nucleoside and template: for instance, 1D binds to 85% binding sites available on a20, and even 100% binding sites on A20 are occupied by 2L.
| Template | 1D | 1D | 1L | 1L | 2D | 2D | 2L | 2L |
|---|---|---|---|---|---|---|---|---|
| f (%) | c | f (%) | c | f (%) | c | f (%) | c | |
| A20 | 90 ± 9 | − | 65 ± 7 | − | 100 ± 10 | − | 100 ± 10 | + |
| a20 | 85 ± 9 | + | 80 ± 8 | + | 85 ± 9 | − | 100 ± 10 | + |
These results conclusively show that there is no (or at least no significant) configuration-selective recognition between these four nucleosides and their templates A20 and a20. The DNA assemblies with 1D and 1L show only slightly reduced occupancy rates if their configuration does not match with that of the template. A closer look on the absorbance reveal also only very minor alterations indicating also only slight differences for stacking interactions. Based on the knowledge that the new L-configured nucleoside conjugates 1L and 2L bind to both A20 and a20 the specific base pairing was additionally checked with T20, G20 and C20. In contrast to A20 (and a20), the three “wrong” DNA templates T20, G20 and C20 are not able to keep a significant amount of 1L or 2L in aqueous solution (see Fig. S20 and S21, ESI†). This shows that the binding of 1L and 2L to the DNA templates follows the specific base pairing rules as previously evidenced for 1D and 2D assembled along A20.20 Similar to double-stranded DNA, these chromophore assemblies show cooperativity with a melting temperature at approximately 70 °C (see ESI,† for 2D with A20, Fig. S26).
1D shows eximer-like fluorescence in DNA-templated assemblies, whereas the fluorescence of 2D is completely quenched in the assemblies.20 Hence, we did not further investigate the fluorescence properties of the other chiralities. More importantly, we probed the chromophore assemblies bound to the DNA templates (after centrifugation) by circular dichroism (CD) spectroscopy in order to gain more information on the helical chromophore chirality. All four applied chromophore–nucleoside monomers alone show no significant CD signal, which is typical for small molecules, although they bear the chiral 2′-deoxyribofuranosides. Moreover, unbound aggregates have been removed by centrifugation during the sample preparation. Taken together, CD spectroscopy selectively probes the assemblies of chromophores along the DNA templates without overlaying signals from potentially present excess, but unbound monomers. Unfortunately, the helicity of the DNA core consisting of the 2′-deoxyadenosines of the templates and the 2′-deoxyuridines of the chromophore conjugates 1D, 1L, 2D and 2L in these supramolecular assemblies cannot be clearly determined, because the CD spectra in the absorption range of the A-dU base pair core around 260–280 nm is overlaid by the ethynyl pyrene or ethynyl nile red chromophores that also absorb light in this range. But the CD spectra of the ethynylpyrene-2′-deoxyuridine assemblies (Fig. 2) show clear exciton-coupled signals in the typical ethynylpyrene absorption range with two Cotton effects at 370 nm and 413 nm and an intervening axis intersection at 385 nm. The assembly of 1D along A20 serves as first reference and shows positive Cotton effect followed by a negative one. This is opposite to the published CD spectrum of a right-handed double-stranded DNA with five covalently attached building blocks of 1D.27 Accordingly, the non-covalent assembly of 1D along A20 can be assigned to a left-handed chromophore helicity. Interestingly, such left-handed chirality was also found by others for binding of Zn(II)-cyclen perylene/naphthalenebisimide conjugates to the same template A20 indicating complexer mechanisms of assembly.28 Moreover, the assembly of 1L along A20 shows also left-handed helicity, although the configuration of the nucleoside and the DNA template do not match. In contrast, both assemblies of 1D and 1L, each along a20, show opposite CD effect and thus right-handed helicity. The CD signal of 1L with A20 is weaker probably than the others due to the lower occupation fraction of 65% in this assembly. This makes conclusively clear, that the chirality of the assembled supramolecular chromophore helix is controlled by the configuration of the DNA template: The D-configured template A20 yields left-handed helical ethynyl pyrene assemblies whereas the L-configured a20 yields right-handed helical assemblies.
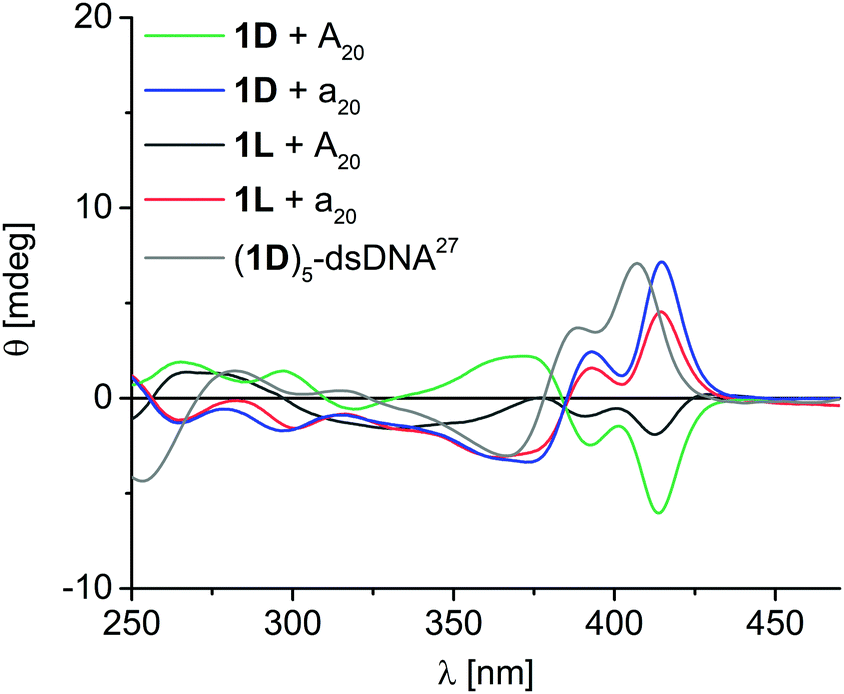 | ||
| Fig. 2 Circular dichroism spectra of DNA assemblies with 1D and 1L along A20 and a20; [DNA template] = 1.25 μM, [nucleosides] = 25 μM in water + 2% DMSO, supernatant after centrifugation; grey: double-stranded (ds) DNA covalently modified with five units of 1D adjacent to each other (2.5 μM in 50 mM Na–Pi buffer, 250 mM NaCl, pH 7.0, r.t.).27 | ||
For the assemblies with 2D and 2L exciton-coupled CD signals were observed in the ethynyl nile red-typical absorption range, consisting of two Cotton effects at 517 nm and 566 nm, and an intervening axis intersection at 540 nm (Fig. 3). The comparison of the CD of the 2D assembly along A20 with the published CD of a DNA with five covalently attached 2D building blocks,29 reveals again left-handed chirality. However, in contrast to 1D and 1L, the chirality of the DNA assemblies with 2D and 2L is not at all controlled by the configuration of the DNA template. Instead, both assemblies of 2D along A20 (matching) and along a20 (non-matching configuration) show left-handed helicity, whereas both assemblies of 2L along A20 and a20 show right-handed helicity. Obviously, the stronger π–π-stacking interactions between ethynyl nile red chromophores that gain not only higher occupancy fractions at the templates additionally control the chirality of the helical DNA assemblies. It is known that 2D stacks in left-handed chiral aggregates even in the absence of any template29 and thereby overrules the chirality control by the templates A20 and a20 that was observed for 1D and 1L. This result tracks well with an important observation by Schenning et al., that, for larger π–π interactions between chromophores in the assembly, the hydrogen bonding interaction to the template diminishes, and the stacking has stronger influence on the structure, here in particular chirality.30,31
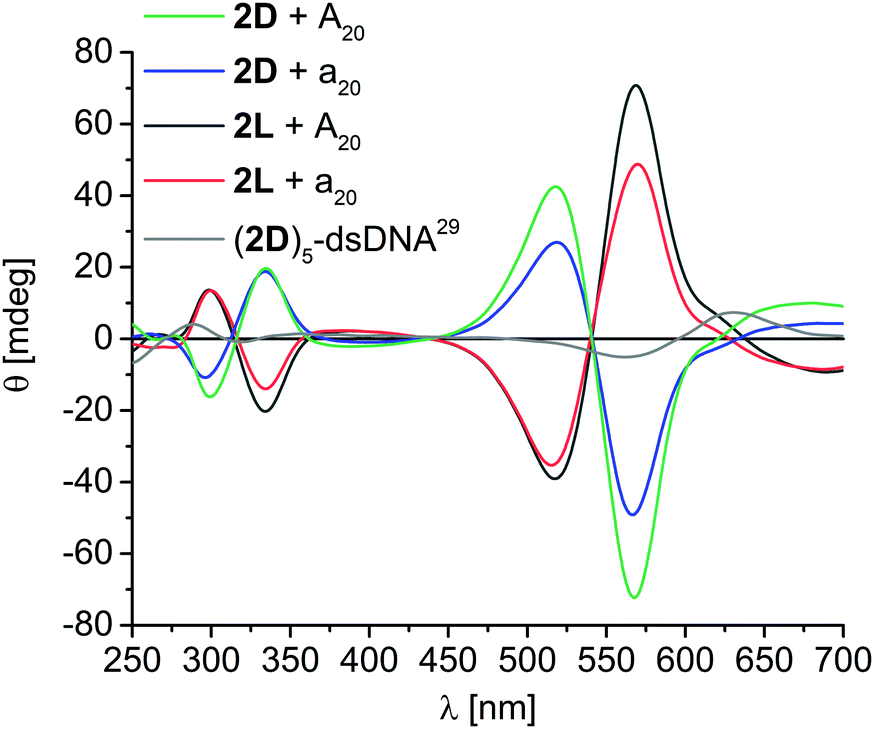 | ||
| Fig. 3 Circular dichroism spectra of DNA assemblies with 2D and 2L along A20 and a20 templates; [DNA template] = 1.25 μM, [nucleosides] = 25 μM in water + 2% DMSO, supernatant after centrifugation; grey: double-stranded (ds) DNA covalently modified with five units of 2D adjacent to each other (1.0 μM in 50 mM Na–Pi buffer, 250 mM NaCl, pH 7.0, r.t.).29 | ||
In conclusion, we studied the way how chirality is controlled in supramolecular DNA-templated chromophore assemblies. We applied a combination of four different D- and L-configured chromophor–nucleoside conjugates, 1D/1L and 2D/2L, to probe their non-covalent assembly along the D- and L-configured templates A20 and a20, respectively. There are several results: (i) the chirality of these supramolecular chromophore–DNA architectures is not simply controlled by the configuration of the DNA template, which could have been expected based on the published D-/L-DNA.25,26 (ii) Although the selective binding of the chromophore–2′-deoxyuridine conjugates follow specific base pairing rules, there is no configuration-selective recognition by the DNA templates. 1D and 2D form not only helical assemblies with their configuration-matching template A20 but also with the non-matching a20. (iii) The helicity of the DNA assemblies consisting of the ethynylpyrene conjugates 1D and 1L are controlled by the configuration of the DNA template. Interestingly, the chirality of these non-covalent assemblies is opposite to the published covalently connected DNA architectures.27 (iv) In contrast, the ethynyl nile red conjugates 2D and 2L form chiral stacks (as previously evidenced also for non-DNA-templated stacks of 2D)29 and thus control the chirality of their DNA-templated assemblies. They overrule the control by the configuration of the DNA template, presumably by their stronger π–π-stacking interactions. Taken together, these are important and basic results for the design of DNA-architectures, in particular with respect to their application as emitters and sensors for circularily polarized luminescence.
Financial support by the Deutsche Forschungsgemeinschaft (grant Wa 1386/20-1), the Karlsruhe School of Optics and Photonics (KSOP), and KIT is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- Y. Ke, Curr. Opin. Struct. Biol., 2014, 27, 122–128 CrossRef CAS PubMed.
- B. Sacca and C. M. Niemeyer, Angew. Chem., Int. Ed., 2012, 51, 58–66 CrossRef CAS PubMed.
- F. Zhang, J. Nangreave, Y. Liu and H. Yan, J. Am. Chem. Soc., 2014, 136, 11198–11211 CrossRef CAS PubMed.
- R. Varghese and H.-A. Wagenknecht, Chem. Commun., 2009, 2615–2624 RSC.
- E. Stulz, Acc. Chem. Res., 2017, 50, 823–831 CrossRef CAS PubMed.
- C. D. Bösch, S. M. Langenegger and R. Häner, Angew. Chem., Int. Ed., 2016, 55, 9961–9964 CrossRef PubMed.
- P. K. Dutta, S. Levenberg, A. Loskutov, D. Jun, R. Saer, J. T. Beatty, S. Lin, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2014, 136, 16618–16625 CrossRef CAS PubMed.
- M. Surin, Polym. Chem., 2016, 7, 4137–4150 RSC.
- T. F. A. D. Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef PubMed.
- A. Ruiz-Carretero, P. G. A. Janssen, A. L. Stevens, M. Surin, L. M. Herz and A. P. H. J. Schenning, Chem. Commun., 2011, 47, 884–886 RSC.
- P. G. A. Janssen, J. Vandenbergh, J. L. J. V. Dongen, E. W. Meijer and A. P. H. J. Schenning, J. Am. Chem. Soc., 2007, 129, 6078–6079 CrossRef CAS PubMed.
- R. Iwaura, F. J. M. Hoeben, M. Masuda, A. P. H. J. Schenning, W. W. Meijer and T. Shimizu, J. Am. Chem. Soc., 2006, 128, 13298–13304 CrossRef CAS PubMed.
- G. Sargsyan, B. M. Leonard, J. Kubelka and M. Balaz, Chem. – Eur. J., 2014, 20, 1878–1892 CrossRef CAS PubMed.
- G. Sargsyan, A. A. Schatz, J. Kubelka and M. Balaz, Chem. Commun., 2013, 49, 1020–1022 RSC.
- Y. Vyborna, M. Vyborni and R. Häner, Chem. Commun., 2017, 53, 5179–5181 RSC.
- P. Ensslen, F. Brandl, S. Sezi, R. Varghese, R.-J. Kutta, B. Dick and H.-A. Wagenknecht, Chem. – Eur. J., 2015, 21, 9349–9354 CrossRef CAS PubMed.
- S. Sezi and H.-A. Wagenknecht, Chem. Commun., 2013, 49, 9257–9259 RSC.
- P. Ensslen, Y. Fritz and H.-A. Wagenknecht, Org. Biomol. Chem., 2015, 13, 487–492 RSC.
- P. Ensslen, S. Gärtner, K. Glaser, A. Colsmann and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2016, 55, 1904–1908 CrossRef CAS PubMed.
- R. Hofsäβ, S. Sinn, F. Biedermann and H.-A. Wagenknecht, Chem. – Eur. J., 2018, 24, 16257–16261 CrossRef PubMed.
- I. Kocsis, A. Rotaru, Y.-M. Legrand, I. Grosu and M. Barboiu, Chem. Commun., 2016, 52, 386–389 RSC.
- R. Varghese and H.-A. Wagenknecht, Chem. – Eur. J., 2009, 15, 9307–9310 CrossRef CAS PubMed.
- H. Urata, E. Ogura, K. Shinohara, Y. Ueda and M. Akagi, Nucleic Acids Res., 1992, 20, 3325–3332 CrossRef CAS PubMed.
- H. Urata, K. Shinohara, E. Ogura, Y. Ueda and M. Akagi, J. Am. Chem. Soc., 1991, 113, 8174–8175 CrossRef CAS.
- J. Barbaric and H.-A. Wagenknecht, Org. Biomol. Chem., 2006, 4, 2088–2090 RSC.
- J. Rubio-Magnieto, M. Kumar, P. Brocorens, J. Idé, S. J. George, R. Lazzaroni and M. Surin, Chem. Commun., 2016, 52, 13873–13876 RSC.
- R. Varghese and H.-A. Wagenknecht, Chem. – Eur. J., 2010, 16, 9040–9046 CrossRef CAS.
- M. Surin, P. G. A. Janssen, R. Lazzaroni, P. Leclere, E. W. Meijer and A. P. H. J. Schenning, Adv. Mater., 2009, 21, 1126–1130 CrossRef CAS.
- D. Paolantoni, J. Rubio-Magnieto, S. Cantel, J. Martinez, P. Dumy, M. Surin and S. Ulrich, Chem. Commun., 2014, 50, 14257–14260 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of 1L, 2L and a20, and additional optical spectroscopy. See DOI: 10.1039/c8cc08887j |
| This journal is © The Royal Society of Chemistry 2019 |