
Organic chelate-free and azido-rich metal clusters and coordination polymers from the use of Me3SiN3: a new synthetic route to complexes with beautiful structures and diverse magnetic properties
Theocharis C.
Stamatatos
 *ab and
Eva
Rentschler
*b
*ab and
Eva
Rentschler
*b
aDepartment of Chemistry, 1812 Sir Isaac Brock Way, Brock University, L2S 3A1 St. Catharines, Ontario, Canada. E-mail: tstamatatos@brocku.ca; Tel: +1 9056885550 ext. 3400
bInstitute of Inorganic and Analytical Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10-14, D-55128 Mainz, Germany. E-mail: rentschler@uni-mainz.de; Fax: +4961313923922; Tel: +4961313925491
First published on 22nd November 2018
Abstract
In this Feature Article, we highlight the feasibility of a new, recently developed approach towards the synthesis of high-spin molecules and single-molecule magnets (SMMs). The key to the preparation of such molecular compounds is the organic azide precursor Me3SiN3, which fosters the formation of 3d-metal azido clusters and coordination polymers without requiring the assistance of any organic chelating/bridging group. All the isolated compounds contain metallic cores which are surrounded by end-on bridging N3− groups. Consequently, the reported molecular materials exhibit ferromagnetic exchange interactions between the spin carriers, resulting in the stabilization of the maximum possible spin ground states. The additional presence of magnetic anisotropy, depending on the 3d-metal ion used, gives rise to SMM properties with interesting and rare features. This article intends to introduce the readers to an unexplored, but in coordination chemistry very useful azido-based reagent and emphasize the pronounced differences in the structures and magnetic properties of the compounds that result from the reactions between 3d-metals and NaN3 or Me3SiN3 in the presence of organic bridging groups, and 3d-metals with Me3SiN3 alone.
 Theocharis C. Stamatatos | Theocharis C. Stamatatos was born in Patras, Greece, in 1980. He received his BSc degree in Chemistry from the University of Patras (Greece, 2003) and his PhD degree from the same institution (2006), under the supervision of Professor Spyros P. Perlepes. He then completed a postdoctoral appointment at the University of Florida (USA, 2016–2018) with Professor George Christou followed by a one-year military service (2009–2010) in the Hellenic Air Force. He was a temporary Lecturer of Chemistry at the University of Patras (2010–2012), after which he joined the faculty of Brock University (Canada, 2012–2018), where he rose through the ranks to become an Associate Professor of Inorganic Chemistry. He is the author or co-author of over 150 scientific articles and reviews. He has been awarded with the following distinctions: Young Investigator Award (ACS, 2007), MAGMANet European Network of Excellence (Florence, 2008), Chancellor's Chair for Research Excellence (Brock University, 2016), and Humboldt Fellowship for Experienced Researchers (Alexander von Humboldt Foundation, 2016–2018). |
 Eva Rentschler | Eva Rentschler studied chemistry at the Philipps University of Marburg (Germany). After graduating in crystallography from Professor Werner Massa, she completed her doctoral thesis on model complexes of nitrogenase with Professor Kurt Dehnicke. From 1993 to 1997, she was a post-doc in Florence (Italy) with Professor Dante Gatteschi. She started her habilitation thesis in 1997 at the Max Planck Institute in Mülheim and graduated in 2003 at the University of Düsseldorf (Germany). Just a few weeks later she accepted the call for a professorship at the University of Mainz. Since then, she has been teaching and researching new molecular magnetic materials. She is the author or co-author of over 160 scientific articles and reviews. She was awarded scholarships such as the Marie Curie Individual Grant or the Lise Meitner scholarship. Since 2016, she has been a member of the executive board of the Collaborative Research Center/TRR Spin+X. She is repeatedly a post-doc host for the DAAD and the Humboldt Foundation. In 2016 she was elected senior member of the Gutenberg Academy. |
Introduction
Article organization
The main scope of this Feature Article is to highlight the employment of Me3SiN3 in coordination chemistry as a means of obtaining structurally and magnetically interesting cluster compounds and multidimensional coordination polymers with diverse properties. Many recently reported and unpublished examples from our and other research groups are presented in the corresponding subsections, which are organized in terms of the employed metal ion. We tried to avoid repetitive and very extensive discussions about concepts which have already been reviewed by few of us and many others, such as the different strategies to the synthesis of metal clusters and coordination polymers, the use of the azido group as a bridging ligand and a magnetic mediator in inorganic chemistry and magnetism, and the theory beyond high-spin molecules, molecule-based magnets, and single-molecule magnets. We refer the readers to an extensive list of review articles which we think are comprehensive and tutorial enough to provide them with the necessary understanding of the fundamental concepts of those individual research topics. In addition to the discussion of the core research areas in which metal-azido complexes play a pivotal role, we decided to also include in the introduction two small sections devoted to the potential applications of metal-azido complexes in photochemistry and high energy density materials, two research directions that are still growing and worthy of attention. In each of the Results and discussion subsections, emphasis is given on the employed synthetic route, the description of structures, the most interesting magnetic properties of the resulting compounds, and the magnetostructural role of the bridging azido groups.Metal clusters and coordination polymers
Polynuclear metal complexes (or metal clusters or for simplicity clusters) and coordination polymers are 0-D or multidimensional extended structures, respectively, which contain a large number of metal ions that aggregate through a combination of inorganic and/or organic bridging/chelating ligands.1 These molecule-based materials have found applications in many different fields of fundamental and applied research, such as magnetism,2 optics,3 conductivity,4 catalysis,5 materials science,6 nanoscience and nanotechnology,7 bioinorganic chemistry,8 drug delivery,9 sensing and gas storage,10 to name just a few. The key to the preparation of structurally novel molecule-based materials with interesting physicochemical properties is undoubtedly the selected synthetic route.There have been many synthetic approaches developed over the last 30 years,11 but the self-assembly route still remains the most widely used strategy for the crystallization and isolation of these molecular species. Self-assembly is a very diverse and unpredictable synthetic approach, but it is that unpredictability which has allowed synthetic inorganic chemists to prepare, crystallize and study myriads of novel clusters and functional coordination polymers with interesting properties.12 The self-assembly route relies on the reactions of metal ions in different oxidation states with several different classes and types of organic bridging/chelating ligands. The resulting reaction solutions could also be supplemented by smaller in size ancillary bridging ligands, such as carboxylates, inorganic anions, pseudohalides and β-diketones, to assist with the stability and crystallinity of the most thermodynamically or kinetically stable compound.13 The lack of predictability of this approach is further supported by the presence of bridging O2− and/or OR− (R = H, Me, Et) groups, which result from the deprotonation of H2O- or ROH-containing solvents and starting materials or the reduction of atmospheric O2. Oxido, hydroxido and alkoxido bridging groups are often found within the metallic cores and their adopted bridging modes are also unpredictable, depending on the metal oxidation state, the coordination environment, the reaction conditions and the core topologies. From all these, it becomes apparent that the choice of organic chelating and ancillary groups is maybe the most challenging synthetic task in modern coordination chemistry.
High-spin molecules and single-molecule magnets
Within the field of molecular magnetism, high-spin molecules and single-molecule magnets (SMMs) are undoubtedly two areas of research in which polynuclear 3d-metal complexes have played a pivotal role.14 Very high-spin molecules with large S values result from the ferromagnetic exchange interactions between the paramagnetic metal ions that are bridged by organic and/or inorganic ligands. Predominant ferromagnetic coupling is a non-trivial case in metal clusters, given the occasionally very large number of metal ions and ligands involved, and the unpredictability of the nature and strength of the competing magnetic exchange interactions.15 Another difficult – if not impossible – factor to control in metal clusters is the overall magnetic anisotropy, and particularly the zero-field splitting, as defined by the axial parameter, D. The molecular anisotropy is the tensorial sum of the metals’ single-ion values, which are affected by a number of chemical, structural and electronic parameters, such as the crystal and ligand field, the coordination geometry, the spin–orbit coupling, and the spin exchange interactions.16 When non-zero S and negative D values are combined, coordination compounds may exhibit slow magnetization relaxation and SMM behavior.17 The assignment of these properties is often based on the appearance of frequency-dependent out-of-phase (χM′′) signals and hysteresis loops, the diagnostic property of a magnet. Therefore, SMMs have recently been proposed as suitable candidates for emerging technologies including high density data storage, spintronics, and quantum computation.18Azide ions in chemical synthesis and molecular magnetism
The azido group (N3−) is the most commonly employed pseudohalide bridging ligand for the synthesis of 3d-metal clusters and coordination polymers with multiple and tunable physical properties.19 When acting as a monoatomic bridge (end-on coordination mode, EO), the azido ligand can link up to four metal ions. When both terminal nitrogen atoms are used, it can bridge two metal centers (end-to-end coordination mode, EE), while the rare combination of EO and EE modes (EO/EE) is capable of bridging up to six metal ions (Scheme 1).20 The most familiar coordination mode of N3− in cluster chemistry is the EO.21 This allows the bridging azido groups to occupy key sites of the metallic cores and the organic chelates to surround the periphery and support the stability of the core structures. In contrast, EE and EO/EE coordination modes are more frequently found in coordination polymers since these bridging modes foster the aggregation of the metal ions into different dimensions (1-D, 2-D or 3-D).22 Overall, the azide ion is a versatile ligand, meaning that it is generally very difficult to predict what bridging modes will be observed in the resulting compounds.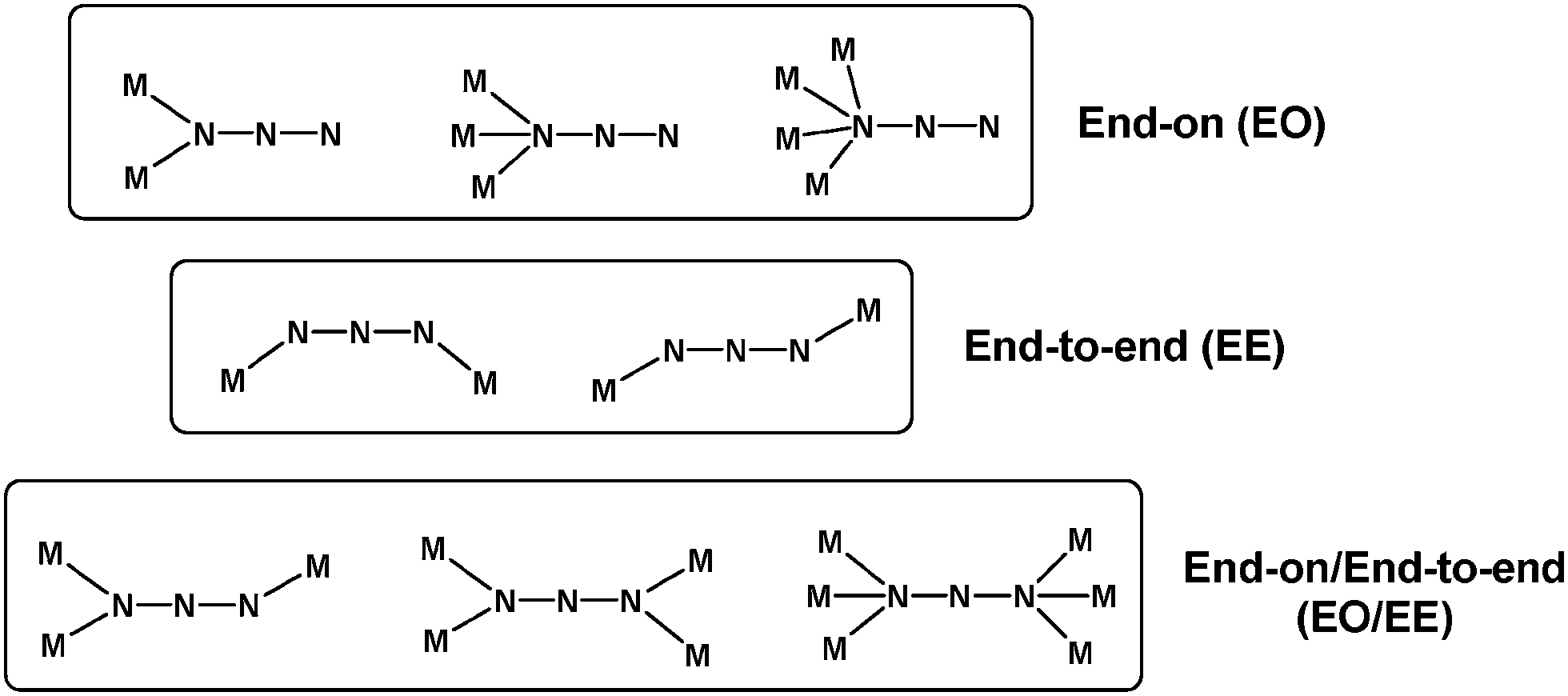 | ||
| Scheme 1 Crystallographically established coordination modes of the bridging azido ligand in coordination chemistry. | ||
Also, in the field of molecular magnetism, the bridging azido group is one of the most popular ligands. In general, EE azido groups propagate antiferromagnetic (AF) interactions, whereas the EO coordination modes are associated with ferromagnetic coupling;23 exceptions to this general rule have been reported.24 However, the bridging mode is not the only factor that affects the nature of the magnetic interactions. Metric parameters such as the bridging/dihedral angles and bond distances and other factors such as the strict and accidental orthogonality of magnetic orbitals, spin polarization and delocalization of unpaired electrons were found to significantly affect the strength and sign of the magnetic coupling between the spin carriers.19,20,22,23,25 Hence, the vast majority of metal-azido clusters and coordination polymers with high-spin values, SMM properties and other interesting magnetic behaviors (i.e., spin flop, canted ferromagnetism, long-range ordering and single-chain magnetism) have been prepared from the self-assembly reactions between metal ions, organic chelates and NaN3. This strategy has led to some of the highest-spin molecular systems to date, including the {Mn19} cluster with S = 83/2,26 the {Mn17} octahedron with S = 37,27 and the {Mn25} barrel-like complex with S = 51/2.28 However, the same synthetic route has also afforded many complexes with very low S values due to the competing interactions between the EO-bridging azides, the organic chelates and the bridging oxido/hydroxido/alkoxido groups.29 Obviously, the main drawback of this approach is the reliability that the resulting complexes will exhibit an appreciable S value, which could subsequently assist in the onset of slow magnetization relaxation and SMM properties.
Although organic azides have been extensively employed in organic synthesis and catalysis for the preparation of several interesting new compounds, such as nitrides, triazoles, tetrazoles, and amines,30 to name a few, they have a very limited use in metal cluster and coordination polymer chemistry (vide infra). From an inorganic chemistry perspective, the organic azido precursor Me3SiN3 has some noticeable differences when compared to its inorganic counterpart, NaN3, which is the most popular azido-based reagent in coordination chemistry. Firstly, Me3SiN3 is more soluble, allowing the performance of inorganic reactions in various organic solvents. Secondly, the non-coordinated Me3Si+ cation renders possible the isolation of alkali metal-free compounds; the alkali metal ions are often found to compete with 3d-metal ions upon coordination with N/O-based ligands.31 Lastly, Me3SiN3 can readily abstract OH− bridging groups from the reaction solution and subsequently release N3− ions for binding to the metal centers. It is worth mentioning at this point that OH−- and N3−-bridging groups have been found to stabilize isostructural, high-nuclearity 3d-metal clusters albeit with completely different magnetic properties. Antiferromagnetically- and ferromagnetically-coupled MII9/OH− and MII9/N3− complexes (MII = Fe,32 Co,33 Ni34) have been reported by the research groups of Perlepes, Escuer and Tuchagues.32–34 Therefore, the complete replacement or removal of hydroxido bridging groups by EO-bridging azides appears to be a fruitful route to high-spin molecules and SMMs.
Photolysis of metal-azido complexes
Wieghardt and co-workers were the first to report the photo-induced cleavage of a N–N bond in a non-heme iron FeIII complex bearing an axial N3− ligand and a redox-innocent macrocyclic ligand occupying the four equatorial sites.35 High-valent FeV intermediates were detected by EPR and Mössbauer spectroscopy. Although the mechanism of photolysis was not totally established, it was believed that the operative channel of N–N cleavage and stabilization of the FeV product were due to intramolecular vibrational energy redistribution, which corresponds to the azide-associated low frequency modes, thus leading to N–N cleavage.36 Moreover, because of the relatively high barrier for rebound between the Fe(V)/N2 and Fe(III)/N3− species, the formed dinitrogen readily escapes the reaction cage.37 Another interesting result was published by Wagner and Nakamoto, where they reported the photoinduced release of N2 from a porphyrin-based FeIII–N3− complex under irradiation with UV-Vis light in frozen CH2Cl2.38 Photoinduced heterolytic cleavage of the N–N bond was accompanied by oxidation of the FeIII center to form the iron nitrido compound [(L)FeV![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N] with concomitant release of N2. The formation of [(L)FeVN] was confirmed by resonance Raman spectroscopy. The two aforementioned examples demonstrate the additional application of transition metal azido complexes in photolysis, as a means of obtaining high-valent metal complexes with the subsequent release of N2.39
N] with concomitant release of N2. The formation of [(L)FeVN] was confirmed by resonance Raman spectroscopy. The two aforementioned examples demonstrate the additional application of transition metal azido complexes in photolysis, as a means of obtaining high-valent metal complexes with the subsequent release of N2.39
Metal-azido complexes as high energy density materials
High energy materials are compounds that store chemical energy.40 They can be either single complexes or a mixture of an oxidizer and fuel elements. Upon stimulation with mechanical, thermal or electrical devices, these materials can undergo rapid decomposition, releasing heat, light, sound and large volumes of gases.41 The amount of energy released depends on the properties of the material, such as the composition, structure, density, heat of formation, and decomposition, to name a few. High energy materials are classified as explosives, propellants and pyrotechnics based on their properties and applications.42 Metal azides belong to the class of primary explosive materials;43 they detonate easily by application of heat, mechanical shocks, sparks, etc. The commonly used metal azides are those consisting of heavy metal ions (i.e., silver, mercury and lead) and the resulting structures exhibit highly symmetrical packing modes, which subsequently lead to high density materials with enhanced properties and applications in both military and non-military contexts.44Much effort has been recently devoted to the synthesis of novel high density energetic compounds which could be more efficient and environmentally benign than the current ones. To this end, transition metal/azido complexes with large N3−/metal ratios appear as very promising “green” candidates, since after decomposition the main product that will be released is the nontoxic dinitrogen (N2) gas.45 The azide ion, the simplest anionic, nitrogen-rich ligand with the highest nitrogen content of 100%, is a prominent energetic species which can increase the heat of formation by about 350 kJ mol−1 and can also be easily used to alter the oxygen balance. The high sensitivity of the already used azido complexes is a clear drawback for the efficiency of these explosive materials.46 However, the search for new simple systems, including metal-azido clusters and coordination polymers with no strongly bound organic molecules, seems to be a very promising and appealing route to a new, unexplored class of high energy density materials.
The following Results and discussion section will be divided into two main parts: (i) the background of Me3SiN3 in coordination chemistry, in conjunction with various organic chelating/bridging ligands, and (ii) the results obtained from a recently developed synthetic approach to purely azido-bridged and organic chelate-free metal clusters and coordination polymers.
Results and discussion
Me3SiN3 in conjunction with organic chelating/bridging ligands
The history of Me3SiN3 in coordination chemistry starts as early as 1995 when Christou and co-workers reported the site-specific ligand variation in the [MnIII3MnIV(μ3-O)3(μ3-Cl)]6+ distorted cubane core, which was proposed as a model for the tetranuclear water oxidation center of Photosystem II.47 The reaction of the preformed cluster [MnIII4O2(O2CMe)6(dbm)2(py)2] (dbmH = dibenzoylmethane) with ∼1.5 equivalents of Me3SiN3 in hot MeCN yielded the complex [MnIII3MnIVO3(N3)(O2CMe)3(dbm)3] (1). The four Mn ions in 1 are linked together through three μ3-O2−, one μ3-1,1,1 end-on bridging azido ligand and three η1:η1:μ acetate groups (Fig. 1). Peripheral ligation about the distorted cubane core is provided by three bidentate chelating dbm− groups. The azido-bridged complex 1 is a member of a large family of similar [MnIII3MnIV(μ3-O)3(μ3-X)]6+ cubanes, where X− = Cl−, Br− and OCN−, bearing different ancillary carboxylate and capping ligands.48 | ||
| Fig. 1 The structure of 1. H atoms have been omitted for clarity. Colour scheme: MnIII purple; MnIV olive green; O red; N blue; C grey. Reproduced from ref. 47 with permission from the Royal Society of Chemistry. | ||
Magnetic susceptibility studies of 1 revealed the presence of predominant ferromagnetic exchange interactions between the metal centers, resulting in an S = 9/2 ground state spin value. The N3− bridging ion has caused some noticeable magnetic changes within the family of [MnIII3MnIV(μ3-O)3(μ3-X)]6+ distorted cubanes with a C3v symmetry (inset of Fig. 2). It was found to weaken the antiferromagnetic MnIII⋯MnIV interaction and strengthen the ferromagnetic MnIII⋯MnIII interaction, thus stabilizing a well-isolated S = 9/2 spin ground state. In addition, magnetization vs. field studies were used to determine the zero-field splitting parameter, D, which was found to be within the |0.3–0.5| cm−1 range. Consequently, complex 1 exhibited frequency dependent out-of-phase (χM′′) signals below 3 K (Fig. 2), characteristic of an SMM with a relatively small energy barrier for the relaxation of magnetization. Complex 1 together with all the members of this family of {Mn4} clusters have been the second family of molecular compounds to function as SMMs, after the pioneer {Mn12} SMMs.49
 | ||
| Fig. 2 Temperature dependent out-of-phase (χM′′) ac magnetic susceptibility signals for 1 at three ac frequencies: (●) 997, (■) 499 and (▲) 250 Hz. The lines are guides for the eye only. (Inset) The [MnIII3MnIV(μ3-O)3(μ3-N3)]6+ distorted cubane core of 1. The color scheme as in Fig. 1. Reproduced from ref. 47 with permission from the Royal Society of Chemistry. | ||
Many years after, and particularly in 2009, Christou with one of us demonstrated for the first time the dual role of Me3SiN3 in not only delivering the desired N3− ions in solution but also acting as a carboxylate abstraction reagent.50 The degree of carboxylate removal (partial vs. complete) by the Me3SiN3 has never been a subject of rational control. However, this synthetic route was proven to be an important advancement in high-oxidation state Mn carboxylate chemistry because it permitted the performance of simple reactions between various low- and high-nuclearity Mn–carboxylate complexes and Me3SiN3 in many different organic solvents. Such a synthetic strategy has never reproduced the same Mn-azido clusters when NaN3 was used in place of Me3SiN3.
To this end, the reaction between the preformed [Mn11O12(O2CPh)15] complex (5MnIII and 6MnIV) and two equivalents of Me3SiN3 in MeCN afforded crystals of the new [Mn25O20(N3)6(O2CPh)26(MeCN)2] (2) cluster in low yields. The structure of 2 (Fig. 3, top) comprises 22 MnIII and 3 MnII atoms bridged by 20, in total, μ3- and μ4-O2− ions to give a nonplanar [Mn25O20]32+ core. The core of 2 can be described as consisting of a central, near-planar {Mn7} unit, which is linked to six additional Mn atoms through the bridging O2− ions. The resulting {Mn13} unit is held within a non-planar (puckered) ring of 12 MnIII atoms by 12 μ3-O2− ions and 6 μ-1,1 (end-on) N3− groups; two of the latter bridge a MnII⋯MnIII pair and four a MnIII⋯MnIII pair. The complex thus contains an overall [Mn25(μ4-O)8(μ3-O)12(μ-N3)6]26+ core (Fig. 3, bottom). Peripheral ligation about this core is provided by 20 η1:η1:μ, 3 η1:η2:μ3, and 3 η2:η2:μ4 bridging PhCO2− groups and two terminal MeCN molecules.
 | ||
| Fig. 3 The structure of 2 (top) and its [Mn25O20(N3)6]26+ core (bottom), highlighting with purple thick lines the outer {Mn12} and inner {Mn6} cyclic units. H atoms have been omitted for clarity. Colour scheme: MnII yellow; MnIII purple; O red; N blue; C grey. Reproduced from ref. 50 with permission from the Royal Society of Chemistry. | ||
The mechanism of {MnIII/IV11} to {MnII3MnIII22} (2) conversion is clearly a very complicated one. However, it is very likely that the carboxylate abstraction, induced by the Me3SiN3, has been associated with the disproportionation of MnIII ions in solution, which would also rationalize the small yield of 2. Nonetheless, we have not isolated any other clean products from this reaction, and there is probably a complicated mixture of species in the filtrate. Magnetic susceptibility studies on complex 2 revealed the presence of predominant antiferromagnetic exchange interactions between the Mn atoms and a resulting S = 3/2 ground state with very low-lying excited states, as expected for such a high-nuclearity complex. It appears that the large number of oxido bridges, a well-known antiferromagnetic mediator in Mn cluster chemistry,51 in the core of 2 has masked the ferromagnetic contributions from the bridging N3− groups. Regardless, complex 2 shows very weak tails of out-of-phase (χM′′) signals below ∼2 K, indicative of a very fast-relaxing SMM with a small barrier.
It was not until recently that we explored comproportionation reactions between MnII and MnVII sources in the presence of both carboxylic acids (mainly as acidifiers to prevent the formation of insoluble Mn oxides) and Me3SiN3. The self-assembly reaction between Mn(NO3)2·6H2O and NBu4nMnO4 in the presence of excess 3,3-dimethylacrylic acid (dmaH) and Me3SiN3 in MeNO2 yielded dark-brown crystals of the complex [Mn29O24(N3)10(dma)28] (3) in moderate yields.52 Complex 3 crystallizes in the highly-symmetric tetragonal space group P4/nnc. Bulk materials such as the important minerals rutile, pyrolusite and zircon have been found to crystallize in similar, high-symmetry space groups.
The structure of 3 (Fig. 4, top) comprises one central MnII ion and 28 MnIII ions held together by 16 μ3- and 8 μ4-O2− ions, as well as 8 μ-1,1 and 2 μ4-1,1,1,1 end-on bridging azido groups to yield an overall [Mn29(μ4-O)8(μ3-O)16(μ4-N3)2(μ-N3)8]28+ core (Fig. 4, bottom) with virtual D4h symmetry. Peripheral ligation about the core is provided by a total of 28 bridging carboxylate groups. The complete core of 3 appears to be organized into three layers. The central layer is a {Mn13(μ4-O)8(μ4-N3)2}20+ unit, which can be described as consisting of eight edge-sharing {MnIIMnIII(μ4-O)} tetrahedra, all with a shared common apex, the MnII atom. The μ4-bridging azido groups act as capping ligands for eight MnIII atoms on the two sides of the {Mn13} unit. Above and below the {Mn13} unit, there are two same {Mn8} layers with a ‘crown’-like motif. These layers comprise a non-linear array of 8 MnIII atoms bridged by 8 μ-O2− groups. The μ-O2− groups are becoming μ3-bridging and – in the additional presence of the 8 μ-N3− groups – they serve to link the three layers together.
 | ||
| Fig. 4 The structure of 3 along the crystallographic c axis (top) and its [Mn29(μ4-O)8(μ3-O)16(μ4-N3)2(μ-N3)8]28+ core (bottom). H atoms have been omitted for clarity. Colour scheme: MnII yellow; MnIII purple; O red; N blue; C grey. Reproduced from ref. 52 with permission from the American Chemical Society. | ||
Although complex 3 contains an appreciable number of end-on bridging azides within its repeating units, the average Mn–N–Mn angle is 111.4°, thus presaging the presence of antiferromagnetic interactions between the respective metal centers. Indeed, the magnetic susceptibility measurements of 3 are suggestive of a small spin ground state, most likely an S = 9/2, which is additionally supported by the concurrent presence of many bridging oxido groups (as in the case of 2). Complex 3 did not exhibit an out-of-phase signal down to 1.8 K, indicating that this is not an SMM. From structural and supramolecular perspectives, the {Mn29} cluster is one of the largest metal clusters reported to date, the first icosa-enneanuclear homometallic cluster compound, and it possesses a 2.2 nm size structure with a high-symmetry spherical topology.
Based on these three examples, the resulting question was obvious: “How possible was it to synthesize molecular compounds bearing exclusively azido bridging ligands”? The answer included several synthetic challenges, and the obvious problems to tackle were mostly related to the solubility and crystallinity of the resulting species. Taking into consideration all these drawbacks, our group initiated a long-term program aiming at the preparation of 3d-metal complexes with diverse magnetic and physicochemical properties using simple metal starting materials and the key reagent Me3SiN3 in the absence of any organic bridging/chelating ligand. The published and some unpublished results which are under further investigation are discussed in the following subsections.
Fe(II/III) and Fe(II) azido polymers and clusters
The results with the redox active metal ion, FeII, rely on the synthetic conditions, and particularly on the presence or absence of O2 from the corresponding reaction systems. Hence, the aerobic reaction of Fe(ClO4)2·xH2O, NEt3, and Me3SiN3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:4 molar ratio in MeCN led to dark orange crystals of the mixed-valence compound [FeIIFeIII(N3)5(MeCN)2]n (4) in good yields.53 The presence of base NEt3 was found to be essential for the isolation of the chelate-free complex 4, as in several other reported 3d-metal/azido compounds (vide infra). Although the mechanism of the formation of clusters and polymers is difficult to rationalize, it is likely that NEt3 increases the pH of the solutions and facilitates the deprotonation of H2O-containing solvents and starting materials to OH− groups. The latter can be then abstracted by the Me3Si+ groups, thus fostering the formation of N3−-rich and OH−-free 3d-metal compounds.
:1:4 molar ratio in MeCN led to dark orange crystals of the mixed-valence compound [FeIIFeIII(N3)5(MeCN)2]n (4) in good yields.53 The presence of base NEt3 was found to be essential for the isolation of the chelate-free complex 4, as in several other reported 3d-metal/azido compounds (vide infra). Although the mechanism of the formation of clusters and polymers is difficult to rationalize, it is likely that NEt3 increases the pH of the solutions and facilitates the deprotonation of H2O-containing solvents and starting materials to OH− groups. The latter can be then abstracted by the Me3Si+ groups, thus fostering the formation of N3−-rich and OH−-free 3d-metal compounds.
Complex 4 is a neutral 2-D coordination polymer (Fig. 5) with the asymmetric unit consisting of two iron centers bridged by five μ-1,1 end-on (EO) azido ligands; peripheral ligation is provided by two terminal MeCN molecules. The oxidation states of the metal ions were based on metric parameters and BVS calculations. As a result, Fe1 and Fe3 atoms are divalent and Fe2 is trivalent, confirming the mixed-valence description for 4. The repeating {FeII2FeIII(N3)7(MeCN)2} unit is bent, with an Fe1⋯Fe2⋯Fe3 angle of 119.1°. Two N3− groups, bound to the central Fe2 atom, become μ-1,1 EO, serving to bridge the {FeII2FeIII(N3)7(MeCN)2} unit with an adjacent one. The Fe2–(μ1,1-N3)2–Fe2′ subunit is strictly planar. The resulting {FeII4FeIII2(N3)14(MeCN)4} moieties are further linked to one another by μ-1,1 EO N3− groups (bound to the ‘nodal’ Fe1 and Fe3 atoms) to finally form aesthetically pleasing [FeII4FeIII6(N3)26(MeCN)8] cyclic units, which create an extended 2-D network of ladder-like hexagonal layers (Fig. 5). The Fe⋯Fe intralayer separations and Fe–(μ1,1-N3)–Fe angles span the range 3.331–3.393 Å and 102.5–106.7°, respectively.
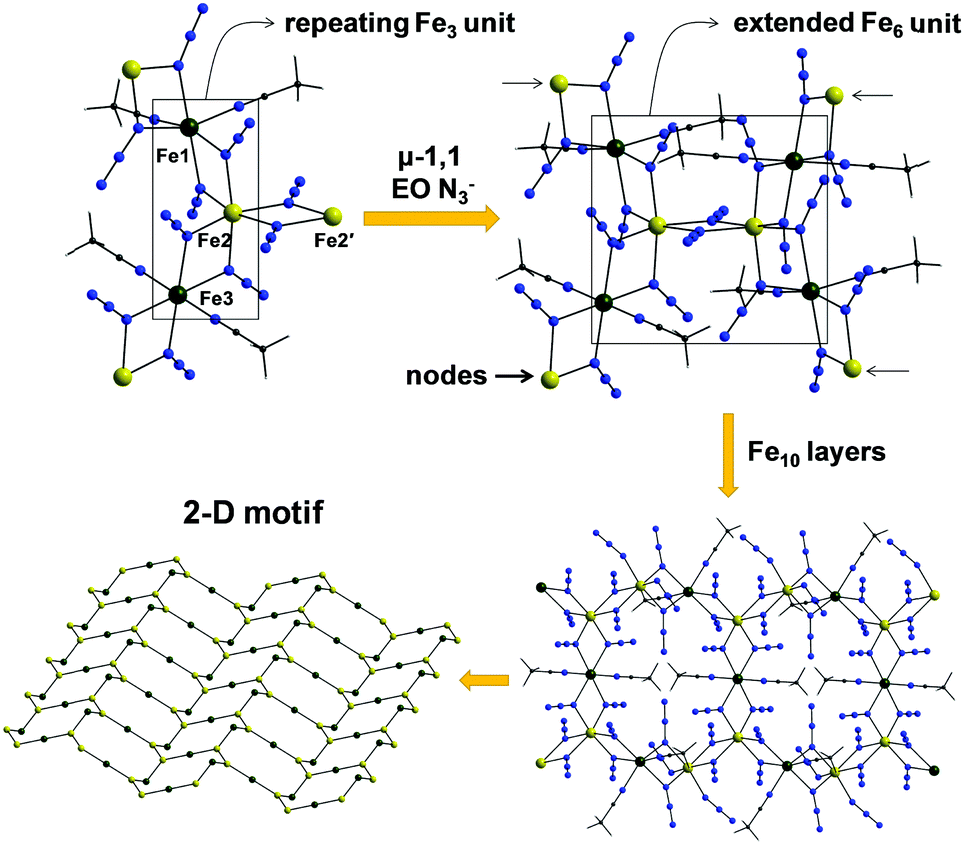 | ||
| Fig. 5 ‘Building up’ the 2-D polymer 4 from simpler fragments. Colour scheme: FeII dark green; FeIII yellow-orange; N blue; C grey; H light grey. Reproduced from ref. 53 with permission from Wiley-VCH. | ||
Magnetic susceptibility studies on a polycrystalline sample of 4 were carried out (Fig. 6, top) and the data in the high-T region (300–80 K) were consistent with the presence of ferromagnetic exchange interactions between the FeII/III centers through the EO N3− bridges. The data above 120 K follow the Curie–Weiss law with θ = +61.0 K, confirming the strong ferromagnetic coupling between the Fe centers. Below 80 K, the χMT product steeply increases, reaching a maximum value at T ∼ 34 K; such behavior is attributed to the long-range ferromagnetic ordering of the spins. The χM and χMT products are strongly field dependent, exhibiting an increase of their values as the field decreases, and thus confirming the ordered phase. To assess the effect of any possible interlayer interactions on the magnetic properties of 4, weaker dc fields (0.002–0.075 T) were also used (inset of Fig. 6). The system retained the same ordering peak although the susceptibility seemed to vanish on cooling when the field was less than 0.002 T. Such behavior is indicative of interlayer interactions and the field to overcome such effects has been estimated to be ∼0.1 T. Finally, the ferromagnetically ordered 2-D system shows magnetization hysteresis loops, still observable at ∼20 K, with a remnant magnetization corresponding to 3.3 electrons and a coercive field of ∼0.25 T at 2 K (Fig. 6, bottom).
 | ||
| Fig. 6 χ M T vs. T plot under a field of 0.1 T (top), field-cooled magnetization plots (inset), and isothermal magnetization plots (bottom) at different temperatures for 4. Reproduced from ref. 53 with permission from Wiley-VCH. | ||
In contrast to the 2-D polymer 4, the anaerobic reaction between divalent Fe(ClO4)2·xH2O, NEt3 and Me3SiN3 in a 1:1:4 ratio in dry MeCN yielded yellow crystals of the heptanuclear 0-D complex [FeII7(N3)12(MeCN)12](ClO4)2·4MeCN (5) in good yields.54 Complex 5, along with the isoskeletal {CoII7} and {NiII7} analogues (see the following subsections), are the first molecular clusters in moderate oxidation states that are exclusively bridged by N3− groups without organic bridging or chelating ligands.
The centrosymmetric heptanuclear cation of compound 5 (Fig. 7, top) consists of a nearly ideal planar hexagon of alternating FeII ions surrounding a central FeII atom, reminiscent of the known Anderson-type structure.55 The {Fe7} disk-like unit possesses virtual S6 symmetry and is stitched together by twelve nitrogen atoms of six μ3-1,1,1 and six μ-1,1 end-on bridging azido ligands. The μ3-1,1,1 azides bridge the {Fe6} hexagon with the central FeII ion, while the μ-1,1 azides link the Fe centers in the outer ring to form the hexagon. Peripheral ligation is completed by twelve terminal MeCN molecules, two on each of the external FeII ions. All FeII ions are six-coordinate with nearly octahedral geometries. The [Fe7(μ3-N3)6(μ-N3)6]2+ inorganic core of 5 can also be described as consisting of six {Fe3(N3)4} partial-cubane units, each double face-sharing and all six vertex-sharing with the central FeII ion. Complex 5 exhibits a layered structure, with layers of N atoms from the azide and MeCN ligands situated above and below the {Fe7} plane.
 | ||
| Fig. 7 The structure of the cation of 5 (top), and a packing diagram of the {Fe7} cations, viewed along the crystallographic c-axis (bottom). Colour scheme: FeII dark green; N blue; C grey; H light grey. Reproduced from ref. 54 with permission from the Royal Society of Chemistry. | ||
Several magnetostructural correlations for azido-bridged systems, especially for CuII, NiII and MnII complexes, have been developed and discussed in terms of the exchange interactions between non-orthogonal magnetic orbitals.56 Importantly, the MII–N3(EO)–MII angles affect the nature and strength of the superexchange interactions. For divalent 3d-metal complexes with EO bridging N3− ligands, the angle for switching from ferro- to antiferromagnetic coupling is typically >104°. Exchange interactions between the FeII ions in 5 (av. Fe–N–Fe angle = 99.2°) were therefore expected to be ferromagnetic on the basis of the structural parameters. Importantly, with respect to the magnetic properties of 5, the packing of the {Fe7} cations in the crystal (Fig. 7, bottom) leads to large separations between FeII ions of neighboring molecules, with the closest intermolecular Fe⋯Fe contact being ∼8 Å owing to the coordinated MeCN molecules.
Apart from the chemical and structural properties of 5, its magnetic properties and dynamics appeared to be the most appealing features. Two forms of 5 were magnetically and spectroscopically studied. The dried form, 5-d, was obtained by drying the crystalline material of 5 under N2 for a maximum of 3 h. The pristine (or wet) form, 5-w, was also studied by immediately transferring and sealing the crystalline material in an NMR tube. Elemental analysis data of 5-d were consistent with the formula {Fe7(N3)12(MeCN)2(ClO4)2} that accounts for the presence of the {Fe7(N3)12}2+ core, two ClO4− anions and only two MeCN molecules, instead of the sixteen that appear in the crystal structure of 5 (including the interstitial MeCN molecules). The solvation/de-solvation effects within 5 were further evidenced by the disappearance of the IR bands of MeCN in 5-d, which were clearly present in the IR spectrum of 5-w (ν = 2280–2310 cm−1).
Direct current (dc) magnetic susceptibility studies on both 5-d and 5-w revealed the presence of predominant ferromagnetic exchange interactions between the metal centers, leading to an S = 14 ground state, the maximum possible value for a ferromagnetically-coupled {FeII7} complex. DFT calculations were also performed and they confirmed that both types of interactions between the Fecentral⋯Feouter and Feouter⋯Feouter spin carriers are ferromagnetic with J values of +3.0 and +3.3 cm−1, respectively. DFT calculations have also evaluated the zero-field splitting parameters, D = −0.2323 cm−1 and E/D = 0.027.
Alternating current (ac) magnetic susceptibility studies were carried out for both 5-d and 5-w. Interestingly, the magnetic dynamics of 5-d and 5-w were found to be completely different, revealing a “Janus”-faced SMM behavior for the dried and pristine forms of the {FeII7} compound attributed to solvation/de-solvation effects from the coordinated MeCN ligands. In particular, the dried form of the {FeII7} compound displays frequency dependent out-of-phase (χM′′) signals, which are entirely resolved, in two different temperature regions (Fig. 8, top). The peak maxima for 5-d in the low-T (1.8–2.6 K) and high-T (2.7–5 K) regimes exhibit similar intensities (inset of Fig. 8) and they do not overlap with each other. This is indicative of two individual relaxation processes, which both appear to be thermally assisted. The data were fit to obtain relaxation times (τ) for the respective ac frequencies, and subsequently two Arrhenius-type plots were constructed. The data were fit to the Arrhenius law [lnτ = lnτ0 + Ueff/kBT], resulting in the parameters Ueff = 43.7(1) K and τ0 = 4.2(1) × 10−10 s, and Ueff = 21.9(2) K and τ0 = 1.1(2) × 10−8 s for the high- and low-temperature regions, respectively. In contrast, 5-w exhibited only one relaxation process below ∼3 K (Fig. 8, bottom), and the resulting fitting parameters were Ueff = 14.1(2) K and τ0 = 3.3(2) × 10−7 s.
 | ||
| Fig. 8 (Top) Out-of-phase (χM′′) ac magnetic susceptibility signals for 5-d. (Inset) χM′′ vs. T plot at a representative ac frequency of 933 Hz showing the two separate relaxation processes in two different T regions for 5-d; the solid blue lines represent fits of the data. (Bottom) Frequency dependent χM′′ signals of 5-w. Reproduced from ref. 54 with permission from the Royal Society of Chemistry. | ||
The presence of two thermally-assisted relaxation processes is a rare phenomenon in polynuclear 3d-metal SMMs and, to the best of our knowledge, has only been previously observed for a few members of the {Mn12} family of SMMs as a result of the ‘Jahn–Teller isomerism’ effect.57 Furthermore, divalent FeII exchange-coupled SMMs are limited to three examples, viz., an {FeII2},58 a family of {FeII4} cubanes,59 and a nonanuclear {FeII9} cluster.32 The unusual magnetic behavior of complex 5 was attributed to the operative solvation/de-solvation effects, which in turn resulted in alterations of the strength of the intermolecular magnetic interactions and the magnitude of the overall magnetic anisotropy of the system due to changes in the crystal field around the outer FeII atoms.
The presence of ClO4− counterions within the structure of 5 prompted us to further investigate the system and perform chemical reactivity studies on the effect of the FeII starting material on the chemical and structural identities of the Fe/N3− products. To this end, the same as in 4 and 5, reaction between FeX2, NEt3 and Me3SiN3 in a 1:1:4 ratio in dry MeCN under anaerobic conditions, but with X = BF4−, afforded orange crystals of the new compound [FeII2(N3)4(MeCN)3]n (6) in very good yields.60 Although 6 appears to have a similar formula to that of 4, the two coordination polymers differ substantially from each other. The asymmetric unit of 6 includes two FeII ions, as confirmed by metric parameters (Fe–N distances > 2.1 Å) and BVS calculations, while the expansion of the asymmetric unit revealed an overall 1-D chain of repeating, centrosymmetric [FeII4(N3)8(MeCN)6] units (Fig. 9). The latter repeating unit can be described as a planar {Fe4} rhombus (or an {Fe4} butterfly) with the ‘body’ FeII atoms of the butterfly bridged by two μ3-1,1,1 end-on azido groups. The μ3-1,1,1 N3− and four additional μ-1,1 end-on azido groups serve to link the ‘body’ FeII atoms with the two ‘wing-tip’ FeII sites. Peripheral ligation is provided by six terminal MeCN molecules. All FeII ions are six-coordinate with nearly octahedral geometries. The [FeII4(N3)8(MeCN)6] units are linked to their neighboring units through two μ-1,1 end-on N3− groups to finally give an isolated, almost linear 1-D chain that extends along the crystallographic c-axis. The shortest Fe⋯Fe separation between the 1-D chains is 8.22 Å.
 | ||
| Fig. 9 Section of the 1-D chain of 6, highlighting with a yellow color a representative repeating {Fe4} unit. H atoms have been omitted for clarity. Colour scheme: FeII dark green; N blue; C grey. | ||
The magnetic behavior of 6 shows similar characteristics to that of 4. Briefly, the χMT vs. T plot in the high-T region (300–100 K) indicates the presence of ferromagnetic exchange interactions between the FeII centers through the EO N3− bridges. Below ∼100 K, the χMT product rapidly increases, reaching a maximum value at T ∼ 20 K; such behavior is suggestive of long-range ferromagnetic ordering of the spins (Fig. 10, top). As in 4, the χM and χMT products of 6 are field dependent, and magnetization hysteresis loops are observed below ∼5 K. The thermal dependence of the ac susceptibility of 6 was also recorded at various frequencies (Fig. 10, bottom). Under zero dc field, as well as under different static fields of 0.05 and 0.1 T, the χM′′ components exhibit resolved peaks that correspond to an ordered system.
 | ||
| Fig. 10 (top) χMT vs. T plot of 6 under 0.1 T; the inset shows a zoom-in section of the χMT vs. T plot in the high-T region. (bottom) Thermal dependence of the χM′′ component of 6 at different ac frequencies and under a static dc field of 0.05 T. | ||
Co(II) clusters
The first CoII/N3− compound without organic chelating or bridging ligands was prepared from the 1:1:4 reaction of Co(ClO4)2·6H2O, NEt3, and Me3SiN3 in MeCN. The resulting solution afforded pink crystals of [CoII7(N3)12(MeCN)12](ClO4)2 (7) in high yields via standard crystallization procedures.53 Complex 7 has a similar structure to its {FeII7} analogue 4 (Fig. 11). However, there are some noticeable differences, which are worthy of further discussion. Firstly, the crystals of 7 appeared to be stable at room temperature and no degradation was observed for at least 24 h of exposure to air. Secondly, the magnetic properties of the wet- and dried-forms of 7 were superimposed for both the static and dynamic measurements.
 | ||
| Fig. 11 The structure of the cation of 7. Colour scheme: CoII tyrian purple; N blue; C grey; H light grey. Reproduced from ref. 53 with permission from Wiley-VCH. | ||
Under a dc field of 0.1 T, the value of χMT for 7 at 300 K is much higher than the spin-only value of seven high-spin, S = 3/2, CoII ions, indicating significant orbital contributions of the distorted octahedral CoII ions. Upon cooling, the χMT product rapidly increases to a maximum of 74.53 cm3 mol−1 K at 14 K and then decreases sharply to a value of 46.79 cm3 mol−1 K at 2 K (Fig. 12, top). The fit of the χMT vs. T data in the high-T regime (blue line in Fig. 12) gave an average Jav = +9.34 cm−1 with each SCo = 3/2 and g = 2.50. For the low-T region (2–40 K), a system with effective spins of SCo′ = 1/2 was considered; an excellent fit (red line in Fig. 12) was obtained with Jav = +29.4 cm−1, zJ′ = −0.11 cm−1 and g = 6.63, where zJ′ accounts for the presence of intermolecular magnetic interactions. The fit parameters confirmed the strong intracluster ferromagnetic interactions in 7, which led to an effective spin ground state of S = 7/2. The field-dependent magnetization studies were also performed; the isofield lines were far from superposition, indicating significant magnetic anisotropy. An excellent fit of the reduced magnetization data was obtained assuming a well-isolated, S = 7/2 ground state with a negative D = −5.55 cm−1 and g = 6.56. Consequently, complex 7 exhibited frequency-dependent out-of-phase ac signals below 6 K under zero applied dc field; however, no peak maxima were observed for most of the frequencies down to 1.8 K. The application of a small external dc field of 0.1 T resulted in the suppression of the fast quantum tunneling process, and subsequently an appreciable shift of the out-of-phase ac signals to higher temperatures was observed (Fig. 12, bottom left). Therefore, the peak maxima were clearly detected for all ac frequencies and an Arrhenius plot was constructed from the χM′′ vs. T data (Fig. 12, bottom right). The fit to the thermally activated region gave Ueff = 28.1 K and τ0 = 8.0 × 10−8 s, where τ0 is the pre-exponential factor.
 | ||
| Fig. 12 (top) χMT vs. T plot for 7 under a field of 0.1 T; the solid lines are the fits of the data and the fit parameters are reported in the text. (bottom, left) Out-of-phase (χM′′) vs. T ac magnetic susceptibility signals for 7 under a 0.1 T static dc field, with a 4.0 G ac field oscillating at various frequencies, and (bottom, right) the Arrhenius plot from the peak maxima. Reproduced from ref. 53 with permission from Wiley-VCH. | ||
Compared to all the previously reported {CoII7} disk-like clusters bearing various organic bridging/chelating ligands,61 complex 7 (i) is a stronger ferromagnetic system due to the presence of μ3-1,1,1 and μ-1,1 end-on bridging N3− ions with an average Co–N–Co angle of 96.1°, (ii) has a very well isolated ground state spin value, as evidenced by similar EPR spectra at 4 and 23 K, and (iii) is a better field-induced SMM with an energy barrier amongst the highest yet reported for any polynuclear CoII system.62
Although – strictly speaking – the following example does not fall into the direct scope of this subsection, we felt it timely to report its synthesis and structure in order to highlight the negative effect of the ancillary bridging ligands on the magnetic properties of azido-bridged CoII clusters. Therefore, the reaction between the [Co2(H2O)(piv)4(pivH)4] (pivH = pivalic acid) precursor and excess Me3SiN3 in MeCN afforded purple crystals of the [CoII8(N3)6(piv)12](OH)2 (8) compound in good yields.63 The highly-symmetric structure of 8 consists of eight CoII ions arranged in a cube-like topology (Fig. 13, top). The edges of the {Co8} cube are capped by six μ4-1,1,1,1 end-on azido groups and twelve bidentate η1:η1:μ-bridging piv− ligands. Although the average Co–N–Co angle in 8 is 99°, below the cutoff angle of 104° for the ferro- to antiferromagnetic switching, the presence of bridging carboxylate groups appears to be responsible for the overall antiferromagnetic response and the observed S = 0 spin ground state (Fig. 13, bottom).
 | ||
| Fig. 13 (top) View of the {Co8} structure of 8 and its cubic-like metal topology. H atoms have been omitted for clarity. Colour scheme: CoII tyrian purple; N blue; O red; C grey. (bottom) χMT vs. T plot of 8 under 0.1 T. | ||
Ni(II) clusters
Our first synthetic efforts in NiII/N3− chemistry have led us to the preparation of complex [NiII7(N3)12(MeCN)12](ClO4)2 (9) as green plate-like crystals in very large yields.53 Compound 9 is isomorphous to the {FeII7} (5) and {CoII7} (7) clusters and it possesses a disk-like topology. Similar to 5 and 7, the {Ni7} cluster is also ferromagnetically-coupled with a resulting S = 7 spin ground state. The fit of the χMT vs. T data over the entire temperature region (300–2 K) gave the parameters Jintra = +33.4 cm−1, Jinter = +20.0 cm−1, zJ′ = −0.02 cm−1 and g = 2.23 (Fig. 14). This confirms the strong intramolecular ferromagnetic interactions between the NiII centers, with the first S = 6 excited state lying ∼60 cm−1 above the ground state. This is clearly due to the exclusive presence of EO bridging azido groups, which promote strong ferromagnetic interactions without diminishing their “ferro-coupling potency”, as is usually the case in cluster compounds when bridging azides co-exist with other bridging organic or inorganic (i.e., O2− and OH−) groups. The magnetization of the system reaches a saturated value close to 15M/NμB, which is consistent with an S = 7 ground state and a negligible magnetoanisotropy. As a result, complex 9 does not exhibit out-of-phase ac signals down to 1.8 K, in either the absence or the presence of an external dc field.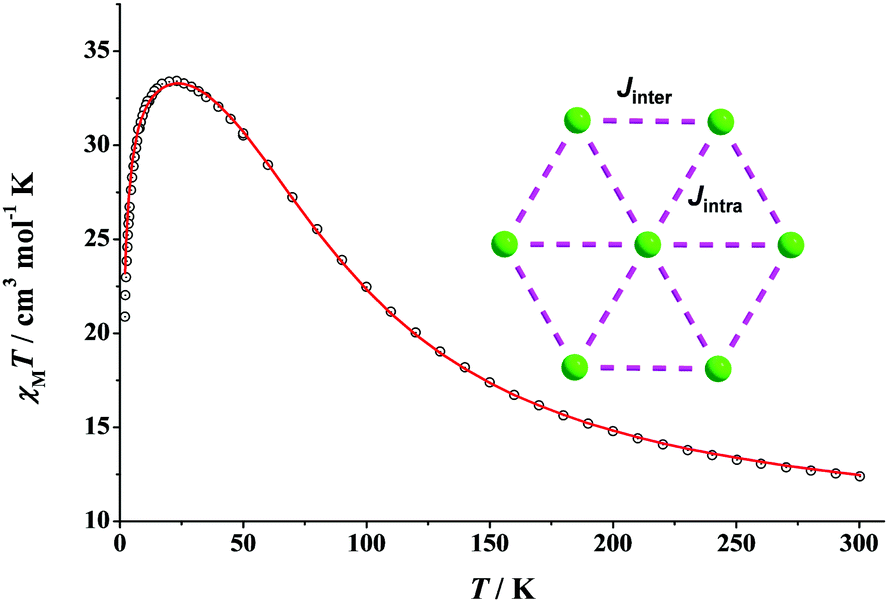 | ||
| Fig. 14 χ M T vs. T plot of 9 under a 0.1 T field. The red solid line is the fit of the data; see the text for the fit parameters. (Inset) 2-J coupling scheme for 9; NiII atoms are shown in green colour. Reproduced from ref. 53 with permission from Wiley-VCH. | ||
Since the isolation of the {Ni7} cluster, we sought ways to overcome the stability of that core structure and prepare NiII-azido clusters with higher nuclearities and possibly larger S values. To this end, we concentrated on the investigation of the size effect of the counterion on the chemical and structural identities of the resulting metal-azido clusters. Among many different anions and anionic species used in place of ClO4− ions, we were recently able to synthesize a new family of cationic [Ni10(N3)18(MeCN)14]2+ clusters counterbalanced by the [Ln(NO3)5]2− (Ln = lanthanides) species.64 Under various synthetic conditions, it appears that only the [Ln(NO3)5]2− complexes were able to template the formation of the extended decanuclear core.
The general reaction of NiII carboxylate starting materials with Ln(NO3)3 in a large excess of Me3SiN3 in MeCN led to the complexes [Ni10(N3)18(MeCN)14][Ln(NO3)5] (LnIII = Gd (10), Tb (11), Dy (12), Y (13)) in yields >60%. The [Ni10(N3)18(MeCN)14]2+ cation is the largest metal cluster in moderate oxidation state that is exclusively bridged by N3− groups. Furthermore, the 1.8:1 N3−/NiII ratio of the {Ni10(N3)18}2+ core is – to our knowledge – the highest ratio reported to date for any high-nuclearity system, potentially making these species attractive candidates for high energy density materials. All complexes 10–13 contain the same [Ni10(N3)18(MeCN)14]2+ cluster cation (Fig. 15, top) with very similar Ni⋯Ni distances and Ni–N–Ni angles. The av. Ni–N–Ni angles for complexes 10, 11, 12 and 13 are 98.4, 98.5, 98.7 and 98.3°, respectively, thus presaging the presence of ferromagnetic exchange interactions within the {Ni10(N3)18}2+ core.
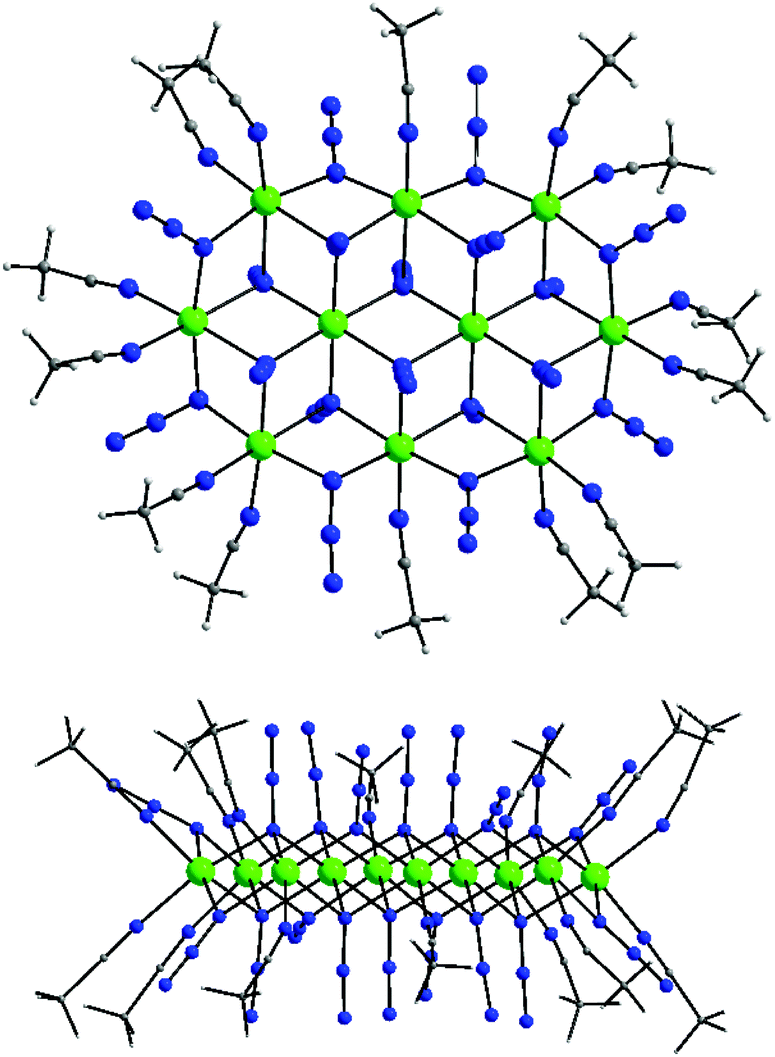 | ||
| Fig. 15 Structure (top) and a side view (bottom) of the {Ni10}2+ cation of complexes 10–13. Colour scheme: NiII green; N blue; C grey; H light grey. Reproduced from ref. 64 with permission from the Royal Society of Chemistry. | ||
The [Ni10(N3)18(MeCN)14]2+ cation can be described as a nearly ideal planar disk-like unit resulting from the fusion of two smaller {Ni7} disk-like moieties. Interestingly, the {Ni10} molecular topology resembles to some extent the structure of the brucite mineral.65 The eight external NiII atoms are bridged by eight μ-1,1 end-on bridging azido ligands, while the linkage between the external and the two central NiII atoms is provided by ten μ3-1,1,1 end-on azides. Peripheral ligation is completed by fourteen terminal MeCN molecules, all on the external NiII atoms. All NiII atoms are six-coordinate with near-octahedral geometries. As in the heptanuclear disk-like complexes, the decanuclear 10–13 have a layered structure, with layers of N-atoms from azides and MeCN above and below the {Ni10} plane (Fig. 15, bottom). All LnIII atoms in 10–13 are 10-coordinate and they are surrounded by ten O-atoms from five bidentate chelating NO3− groups to give an overall distorted tetradecahedral geometry. The closest distance between the LnIII atom in the [Ln(NO3)5]2− counterions and the {Ni10}2+ cluster cation is ∼9 Å for the Tb- and Dy-analogues. Furthermore, the intermolecular Ln⋯Ln distances between neighboring [Ln(NO3)5]2− counterions span the range ∼15–18 Å, sufficiently large to mediate any significant magnetic communication between the LnIII atoms.
To overcome the problems related to the loss of crystallinity and the partial or complete loss of the MeCN molecules upon exposure of the crystals to room temperature (Fig. 16), the magnetic susceptibility measurements of complexes 10–13 were recorded on wet (as prepared) crystalline samples. The χMT vs. T plots are depicted in Fig. 17, and they all show a similar response indicative of the presence of ferromagnetic exchange interactions between the NiII atoms, as expected from the similar av. Ni–N–Ni angles, added to the magnetic contribution from the isolated [Ln(NO3)5]2− counterions.66 For the {Ni10}{Y} analogue, the spin ground state value was nicely confirmed to be S = 10, the maximum possible value for such a system. The similar behavior of the other members of this family was indicative of a small or even negligible effect of the lanthanide's spin and orbital angular momenta on the dominant magnetic properties of the {Ni10} cluster cation. This was further supported by the lack of any ac signals from the Tb- and Dy-analogues, even in the presence of various external dc fields.
 | ||
| Fig. 16 IR spectra of representative complex 12 as a function of time. The exchange of coordinated/lattice MeCN by H2O molecules is evidenced by the disappearance of the IR bands in the 2250–2350 and 2900–3000 cm−1 regions of the wet-sample and the emergence of a broad band ∼3400 cm−1 for the two indicated dried-samples, respectively. Reproduced from ref. 64 with permission from the Royal Society of Chemistry. | ||
 | ||
| Fig. 17 χ M T vs. T plots of 10–13 under an applied dc field of 0.1 T. Reproduced from ref. 59 with permission from the Royal Society of Chemistry. | ||
The fast increase of magnetization for all complexes, even under low fields, was supportive of the presence of ferromagnetic exchange interactions within the {Ni10} cluster cation. The saturation of magnetization at values of ∼27 and ∼21NμB agrees well with S = 27/2 and 10 spin systems (for g ∼ 2) for 10 and 13, respectively. The large S value of complex 10 prompted us to preliminarily evaluate its potential as a magnetic refrigerant,67 through the determination of the magnetic entropy change, ΔSm, from the magnetization data. The value of −ΔSm increased gradually as ΔH increased and T decreased, to reach a value of 17.4 J kg−1 K−1 at T = 3 K and ΔH = 7 T. This value was very close to the maximum entropy expected from S = 10 and S = 7/2 noninteracting spins, that is, 17.9 J kg−1 K−1.
Cu(II) polymers
The chemistry of CuII with Me3SiN3 has been associated with many soluble species in the corresponding filtrates, which made their crystallization and isolation a very challenging task. However, the reaction between Cu(NO3)2·3H2O, NEt3, and Me3SiN3 in a 1:2:6 molar ratio in MeCN afforded dark green crystals of the 2-D coordination polymer {(NHEt3)[Cu4(N3)9]}n (14) in 35% yield.68 Interestingly, complex 14 is the first compound from this library of azido-bridged clusters and polymers which does not contain any coordinated MeCN molecules. As a result, the crystals of 14 are extremely stable at room temperature and they retain their crystallinity even after 5 days. The NHEt3+ countercations were found to occupy the voids of the crystal lattice, preventing the 2-D sheets from expanding into a 3-D network. The anionic [Cu4(N3)9]− repeating unit of 14 consists of four CuII ions that are bridged by μ-1,1 and μ3-1,1,1 end-on azido groups to give a defective dicubane metal topology, i.e. two cubanes sharing a face and each missing one metal vertex. The {Cu4} units extend to 1-D chains (Fig. 18, top), and they are further linked to adjacent {Cu2(μ-1,1-N3)2} zig-zag chains (Fig. 18, middle) to yield an overall 2-D coordination polymer (Fig. 18, bottom). The linkage between the {Cu4} and {Cu2} units is accomplished by μ3-1,1,3 end-on/end-to-end bridging azides, a unique coordination mode within this family of azido-bridged molecular materials. Finally, the CuII ions are four-, five- and six-coordinate in square-planar, trigonal bipyramidal and Jahn–Teller distorted octahedral geometries, respectively. Preliminary magnetic susceptibility studies on complex 14 revealed the presence of predominant ferromagnetic exchange interactions between the metal centers (Fig. 19). Additional magnetic measurements are currently in progress and the combined results will be published in an upcoming full paper.
 | ||
| Fig. 18 Representation of the {Cu4} (top) and {Cu2} (middle) end-on azido bridged units which lead to the overall 2-D structure (bottom) of polymer 14 through the presence of μ3-1,1,3 end-on/end-to-end bridging azides. Colour scheme: CuII cyan; N blue. | ||
 | ||
| Fig. 19 χ M T vs. T plot of 14 under an applied dc field of 0.1 T. | ||
Finally, given the fact that this synthetic route has only been recently developed, we felt it timely to summarize in Table 1 all the structurally and magnetically characterized metal-azido clusters and polymers obtained from the use of Me3SiN3 in the presence or absence of any ancillary bridging/chelating organic ligand. We hope to engage the readers in this growing database of chemically, structurally and magnetically appealing species, and encourage the synthetic and magnetism communities to enrich this list with new, higher-nuclearity, higher-spin molecules, and more efficient SMMs.
| Formulaa,b | Azido bridging mode(s) | Predominant magnetic exchange interactions | S | U eff (in K) | Ref. |
|---|---|---|---|---|---|
| a Excluding all lattice solvate molecules and counterions. b Abbreviations: n.a. = non-applicable; n.r. = not reported; dbm− = anion of dibenzoylmethane; dma− = anion of 3,3-dimethylacrylic acid; piv− = pivalate ion. | |||||
| [Mn4O3(N3)(O2CMe)3(dbm)3] (1) | μ3-1,1,1 (EO) | Ferromagnetic | 9/2 | n.r. (SMM) | 47 |
| [Mn25O20(N3)6(O2CPh)26(MeCN)2] (2) | μ-1,1 (EO) | Antiferromagnetic | 3/2 | n.r. (SMM) | 50 |
| [Mn29O24(N3)10(dma)28] (3) | μ-1,1 (EO) and μ4-1,1,1,1 (EO) | Antiferromagnetic | 9/2 | Non-SMM | 52 |
| [Fe2(N3)5(MeCN)2]n (4) | μ-1,1 (EO) | Ferromagnetic | n.a. | Non-SMM | 53 |
| [Fe7(N3)12(MeCN)12]2+ (5) | μ-1,1 (EO) and μ3-1,1,1 (EO) | Ferromagnetic | 14 | 43.7/21.9 (dried) and 14.1 (wet) | 54 |
| [Fe2(N3)4(MeCN)3]n (6) | μ-1,1 (EO) and μ3-1,1,1 (EO) | Ferromagnetic | n.a. | Non-SMM | 60 |
| [Co7(N3)12(MeCN)12]2+ (7) | μ-1,1 (EO) and μ3-1,1,1 (EO) | Ferromagnetic | 7/2 | 28.1 (1000 Oe) | 53 |
| [Co8(N3)6(piv)12]2+ (8) | μ4-1,1,1,1 (EO) | Antiferromagnetic | 0 | Non-SMM | 63 |
| [Ni7(N3)12(MeCN)12]2+ (9) | μ-1,1 (EO) and μ3-1,1,1 (EO) | Ferromagnetic | 7 | Non-SMM | 53 |
| [Ni10(N3)18(MeCN)14]2+ (10–13) | μ-1,1 (EO) and μ3-1,1,1 (EO) | Ferromagnetic | 10 | Non-SMM | 64 |
| [Cu4(N3)9]n− (14) | μ-1,1 (EO), μ3-1,1,1 (EO) and μ3-1,1,3 (EO/EE) | Ferromagnetic | n.a. | Non-SMM | 68 |
Conclusions and perspectives
In conclusion, we have shown in this Feature Article that it is indeed possible to prepare new 3d-metal azido clusters and coordination polymers with high-spin S values and SMM properties by avoiding the presence of any organic chelating/bridging ligand and exclusively using the key reagent Me3SiN3. The metallic cores of all the isolated complexes are end-on N3− bridged, which ensures the presence of predominant ferromagnetic exchange interactions between the spin carriers and consequently the stabilization of the maximum S value. In the additional presence of magnetic anisotropy, driven by the employed 3d-metal ion, some of these species also exhibit interesting SMM properties. Furthermore, the azido group clearly demonstrates its bridging versatility and capacity towards 3d-metals by adopting many different modes, i.e., μ-, μ3- and μ4-EO, as well as μ3-EO/EE. The peripheral ligation about the metal-azido cores is almost always completed by terminal, volatile MeCN molecules, which are frequently involved in solvation/de-solvation effects, thus unveiling rare magnetic phenomena, such as the two thermally-assisted relaxation processes of the {FeII7} cluster.69We strongly believe that this new synthetic approach could potentially stimulate the research in metal-azido complexes to enter a new era, by yielding nitrogen-rich molecule-based materials with fairly low molecular weights (when compared to analogous nuclearities but with organic chelates), non-trivial structures and exciting magnetic properties. The reported materials could also find some potential use in photochemistry, catalysis, and high energy density materials. These studies, among various other future research plans, are currently in progress and the results will be reported shortly. Some representative short- and long-term research objectives are listed below:
(a) The development of this synthetic route to more redox-active and paramagnetic metal ions, such as V, Cr, and Mn.
(b) The preparation of [MII10(N3)18(MeCN)14][Ln(NO3)5] analogues using more anisotropic than NiII and higher-spin divalent 3d-metal ions, such as CoII and FeII. This could in principle lead to SMMs with large energy barriers and blocking temperatures.
(c) The discovery of synthetic variables which would assist with the increase of the cluster nuclearities and subsequently the crystallization of new structural motifs.
(d) The use of bulkier and less volatile nitrile-based solvents, such as benzonitrile, as a means of enhancing the stability of the compounds at room temperature and better controlling the solvation/de-solvation effects, and consequently the strength of the intermolecular magnetic interactions.
(e) The replacement of the [Ln(NO3)5]2− counterion in the {Ni10} clusters by the recently reported [ReF6]2− species;70 the latter was found to exhibit interesting SMM properties due to its large inherent magnetic anisotropy.
(f) The possibility of activating the iron-azido complexes in a variety of oxidation states towards oxidative transformations; this will open opportunities in photoredox catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The synthetic work from our groups described in this Feature Article is mainly based on the PhD work of two very talented scientists, Dr Dimitris I. Alexandropoulos (Brock University, Canada) and Dr Jasmin Krause (University of Mainz, Germany). A part of the research described was carried out during the post-doctoral studies of D.I.A. at Texas A&M University (USA). Our work has been carried out in collaboration with several great scientists and established research groups. In particular, the authors wish to thank Prof. Kim R. Dunbar and Dr Kuduva R. Vignesh (Texas A&M University, USA), Prof. Albert Escuer (University of Barcelona, Spain), Dr Luís Cunha-Silva (Universidade do Porto, Portugal), and Dr Luca M. Carrella (University of Mainz, Germany). This research was financially supported by Brock University, NSERC-DG, ERA and the Alexander von Humboldt-Stiftung Foundation in the context of a research fellowship for experienced researchers (to Th. C. S.).Notes and references
- For a recent review, see: C. Papatriantafyllopoulou, E. E. Moushi, G. Christou and A. J. Tasiopoulos, Chem. Soc. Rev., 2016, 45, 1597–1628 RSC.
- R. Clérac and R. E. P. Winpenny, Single-Molecule Magnets and Related Phenomena, in 50 Years of Structure and Bonding – The Anniversary Volume, ed. D. Mingos, Springer, Structure and Bonding, 2016, 172, pp. 35–48 Search PubMed.
- J.-C. G. Bünzli and S. V. Eliseeva, Chem. Sci., 2013, 4, 1939–1949 RSC.
- For example, see: E. Coronado and P. Day, Chem. Rev., 2004, 104, 5419–5448 CrossRef CAS.
- For a recent review, see: K. Griffiths and G. E. Kostakis, Dalton Trans., 2018, 47, 12011–12034 RSC.
- (a) A. Cornia, M. Mannini, P. Sainctavit and R. Sessoli, Chem. Soc. Rev., 2011, 40, 3076–3091 RSC; (b) M. Mannini, F. Bertani, C. Tudisco, L. Malavolti, L. Poggini, K. Misztal, D. Menozzi, A. Motta, E. Otero, P. Ohresser, P. Sainctavit, G. G. Condorelli, E. Dalcanale and R. Sessoli, Nat. Commun., 2014, 5, 4852 Search PubMed.
- For a mini-review, see: G. Christou, Polyhedron, 2005, 24, 2065–2075 CrossRef CAS.
- (a) V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6, 1676–1695 RSC; (b) J. S. Kanady, E. Y. Tsui, M. W. Day and T. A. Agapie, Science, 2011, 333, 733–736 CrossRef CAS; (c) C. Zhang, C. Chen, H. Dong, J.-R. Shen, H. Dau and J. Zhao, Science, 2015, 348, 690–693 CrossRef CAS.
- C. Wang, D. Liu and W. Lin, J. Am. Chem. Soc., 2013, 135, 13222–13234 CrossRef CAS.
- For a recent perspective, see: J. A. Mason, M. Veenstrab and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC , and references therein.
- For a recent book with a collection of related papers, see: Single-Molecule Magnets: Molecular Architectures and Building Blocks for Spintronics, ed. M. Holynska, Wiley-VCH, 2018 Search PubMed.
- For review articles, see: (a) A. J. Tasiopoulos and S. P. Perlepes, Dalton Trans., 2008, 5537–5555 RSC; (b) Th. C. Stamatatos, C. G. Efthymiou, C. C. Stoumpos and S. P. Perlepes, Eur. J. Inorg. Chem., 2009, 3361–3391 CrossRef CAS; (c) E. K. Brechin, Chem. Commun., 2005, 5141–5153 RSC; (d) M. Andruh, Dalton Trans., 2015, 44, 16633–16653 RSC; (e) M. Murrie, Polyhedron, 2018, 150, 1–9 CrossRef CAS.
- (a) R. E. P. Winpenny, Transition Metals in Supramolecular Chemistry; Perspectives in Supramolecular Chemistry, ed. J.-P. Sauvage, Wiley, 1999, vol. 5, pp. 193–223 Search PubMed; (b) R. E. P. Winpenny, J. Chem. Soc., Dalton Trans., 2002, 2, 1–10 RSC; (c) R. E. P. Winpenny, Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, 2004, vol. 7, pp. 125–175 Search PubMed.
- (a) R. Sessoli, D. Gatteschi and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, UK, 2006 Search PubMed; (b) C. J. Milios and R. E. P. Winpenny, Struct. Bonding, 2015, 164, 1–110 CAS; (c) A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117–2121 CrossRef CAS PubMed; (d) M. Manoli, R. Inglis, M. J. Manos, V. Nastopoulos, W. Wernsdorfer, E. K. Brechin and A. J. Tasiopoulos, Angew. Chem. Int. Ed., 2011, 50, 4441–4444 CrossRef CAS; (e) T. Liu, Y. J. Zhang, Z. M. Wang and S. Gao, J. Am. Chem. Soc., 2008, 130, 10500–10501 CrossRef CAS; (f) P. Alborés and E. Rentschler, Angew. Chem., Int. Ed., 2009, 48, 9366–9370 CrossRef PubMed; (g) G. F. S. Whitehead, F. Moro, G. A. Timco, W. Wernsdorfer, S. J. Teat and R. E. P. Winpenny, Angew. Chem. Int. Ed., 2013, 125, 10116–10119 CrossRef.
- (a) D. Maniaki, E. Pilichos and S. P. Perlepes, Front. Chem., 2018, 6, 1–28 CrossRef PubMed; (b) R. Inglis, C. J. Milios, L. F. Jones, S. Piligkos and E. K. Brechin, Chem. Commun., 2012, 48, 181–190 RSC; (c) J. R. Gispert, Coordination Chemistry, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) D. Gatteschi and L. Sorace, J. Solid State Chem., 2001, 159, 253–261 CrossRef CAS; (b) S. Demir, I.-R. Jeon, J. R. Long and T. D. Harris, Coord. Chem. Rev., 2015, 289–290, 149–176 CrossRef CAS; (c) Y.-S. Meng, S.-D. Jiang, B.-W. Wang and S. Gao, Acc. Chem. Res., 2016, 49, 2381–2389 CrossRef CAS PubMed; (d) A. Kumar Bar, C. Pichon and J.-P. Sutter, Coord. Chem. Rev., 2016, 308, 346–380 CrossRef; (e) C. Duboc, Chem. Soc. Rev., 2016, 45, 5834–5847 RSC.
- (a) G. A. Craig and M. Murrie, Chem. Soc. Rev., 2015, 44, 2135–2147 RSC; (b) Several tutorial papers on SMMs are reported in: Molecular Nanomagnets and Related Phenomena, ed. S. Gao, Springer-Verlag, Berlin, 2014 Search PubMed; (c) D. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268–297 CrossRef CAS PubMed; (d) K. S. Pedersen, J. Bendix and R. Clérac, Chem. Commun., 2014, 50, 4396–4415 RSC; (e) Molecular Magnets: Physics and Applications, ed. J. Bartolomé, F. Luis and J. F. Fernández, Springer-Verlag, Berlin, 2014 Search PubMed.
- (a) M. del Carmen Giménez-López, F. Moro, A. La Torre, C. J. Gómez-García, P. D. Brown, J. van Slageren and A. N. Khlobystov, Nat. Commun., 2011, 2, 407 CrossRef PubMed; (b) R. Vincent, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, Nature, 2012, 488, 357–360 CrossRef CAS PubMed; (c) G. Aromí, D. Aguila, P. Gamez, F. Luis and O. Roubeau, Chem. Soc. Rev., 2012, 41, 537–546 RSC.
- (a) J. Ribas, A. Escuer, M. Monfort, R. Vicente, R. Cortes, L. Lezama and T. Rojo, Coord. Chem. Rev., 1999, 193-195, 1027–1068 CrossRef CAS; (b) Y.-F. Zeng, X. Hu, F.-C. Liu and X.-H. Bu, Chem. Soc. Rev., 2009, 38, 469–480 RSC; (c) C. Adhikary and S. Koner, Coord. Chem. Rev., 2010, 254, 2933–2958 CrossRef CAS.
- A. Escuer and G. Aromí, Eur. J. Inorg. Chem., 2006, 4721–4736 CrossRef CAS.
- Th. C. Stamatatos and G. Christou, Inorg. Chem., 2009, 48, 3308–3322 CrossRef CAS PubMed.
- (a) X.-Y. Wang, Z.-M. Wang and S. Gao, Chem. Commun., 2008, 281–294 RSC; (b) S. Mukherjee and P. S. Mukherjee, Acc. Chem. Res., 2013, 46, 2556–2566 CrossRef CAS PubMed.
- A. Escuer, J. Esteban, S. P. Perlepes and Th. C. Stamatatos, Coord. Chem. Rev., 2014, 275, 87–129 CrossRef CAS.
- For representative examples, see: (a) T. K. Maji, P. S. Mukherjee, G. Mostafa, T. Mallah, J. Cano-Boquera and N. R. Chaudhuri, Chem. Commun., 2001, 1012–1013 RSC; (b) C. S. Hong, J. Koo, S.-K. Son, Y. S. Lee, Y.-S. Kim and Y. Do, Chem. – Eur. J., 2001, 7, 4243–4252 CrossRef CAS; (c) A. Escuer, M. Font-Bardia, S. S. Massoud, F. A. Mautner, E. Peñalba, X. Solans and R. Vicente, New J. Chem., 2004, 28, 681–686 RSC; (d) S. Saha, S. Koner, J.-P. Tuchagues, A. K. Boudalis, K.-I. Okamoto, S. Banerjee and D. Mal, Inorg. Chem., 2005, 44, 6379–6385 CrossRef CAS.
- (a) J. Commarmond, P. Plumeré, J.-M. Lehn, Y. Agnus, R. Louis, R. Weiss, O. Kahn and I. Morgensten-Badarau, J. Am. Chem. Soc., 1982, 104, 6330–6340 CrossRef; (b) M. F. Charlot, O. Kahn, M. Chaillet and C. Larrieu, J. Am. Chem. Soc., 1986, 108, 2574–2581 CrossRef CAS; (c) M. A. Aebersold, B. Gillon, O. Plantevin, L. Pardi, O. Kahn, P. Bergerat, I. von Seggern, F. Tuczek, L. Öhrström, A. Grand and E. Lelièvre-Berna, J. Am. Chem. Soc., 1998, 120, 5238–5245 CrossRef CAS; (d) A. Bencini, C. A. Ghilardi, S. Midollini and A. Orlandini, Inorg. Chem., 1989, 28, 1958–1962 CrossRef CAS; (e) A. Escuer, R. Vicente, J. Ribas, M. S. El Fallah, X. Solans and M. Font-Bardia, Inorg. Chem., 1993, 32, 3727–3732 CrossRef CAS; (f) A. Escuer, R. Vicente, M. A. S. Goher and F. A. Mautner, Inorg. Chem., 1996, 35, 6386–6391 CrossRef CAS PubMed; (g) C. Adamo, V. Barone, A. Bencini, F. Totti and I. Ciofini, Inorg. Chem., 1999, 38, 1996–2004 CrossRef CAS; (h) S. S. Tandon, L. K. Thompson, M. E. Manuel and J. N. Bridson, Inorg. Chem., 1994, 33, 5555–5570 CrossRef CAS; (i) L. K. Thompson and S. S. Tandon, Comments Inorg. Chem., 1996, 18, 125–144 CrossRef CAS; (j) L. K. Thompson, S. S. Tandon and M. E. Manuel, Inorg. Chem., 1995, 34, 2356–2366 CrossRef CAS; (k) Th. C. Stamatatos, G. S. Papaefstathiou, L. R. MacGillivray, A. Escuer, R. Vicente, E. Ruiz and S. P. Perlepes, Inorg. Chem., 2007, 46, 8843–8850 CrossRef CAS PubMed; (l) T. K. Karmakar, B. K. Ghosh, A. Usman, H.-K. Fun, E. Riviére, T. Mallah, G. Aromi and S. K. Chandra, Inorg. Chem., 2005, 44, 2391–2399 CrossRef CAS PubMed.
- A. M. Ako, I. J. Hewitt, V. Mereacre, R. Clérac, W. Wernsdorfer, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 4926–4929 CrossRef CAS PubMed.
- E. E. Moushi, Th. C. Stamatatos, W. Wernsdorfer, V. Nastopoulos, G. Christou and A. J. Tasiopoulos, Inorg. Chem., 2009, 48, 5049–5051 CrossRef CAS PubMed.
- M. Murugesu, M. Habrych, W. Wernsdorfer, K. A. Abboud and G. Christou, J. Am. Chem. Soc., 2004, 126, 4766–4767 CrossRef CAS PubMed.
- For example, see: (a) R. T. W. Scott, S. Parsons, M. Murugesu, W. Wernsdorfer, G. Christou and E. K. Brechin, Angew. Chem., Int. Ed., 2005, 44, 6540–6543 CrossRef CAS PubMed; (b) Th. C. Stamatatos, C. P. Raptopoulou, S. P. Perlepes and A. K. Boudalis, Polyhedron, 2011, 30, 3026–3033 CrossRef CAS; (c) Th. C. Stamatatos, K. A. Abboud, W. Wernsdorfer and G. Christou, Angew. Chem., Int. Ed., 2008, 47, 6694–6698 CrossRef CAS.
- For an excellent book, see: Organic Azides: Syntheses and Applications, ed. S. Bräse and K. Banert, Wiley, 2010 Search PubMed.
- For example, see: (a) Th. C. Stamatatos, D. Foguet-Albiol, W. Wernsdorfer, K. A. Abboud and G. Christou, Chem. Commun., 2011, 47, 274–276 RSC; (b) D. I. Alexandropoulos, E. C. Mazarakioti, S. J. Teat and Th. C. Stamatatos, Polyhedron, 2013, 64, 91–98 CrossRef CAS; (c) E. E. Moushi, Th. C. Stamatatos, W. Wernsdorfer, V. Nastopoulos, G. Christou and A. J. Tasiopoulos, Angew. Chem., Int. Ed., 2006, 45, 7722–7725 CrossRef CAS PubMed; (d) E. E. Moushi, C. Lampropoulos, W. Wernsdorfer, V. Nastopoulos, G. Christou and A. J. Tasiopoulos, J. Am. Chem. Soc., 2010, 132, 16146–16155 CrossRef CAS PubMed.
- (a) A. K. Boudalis, Y. Sanakis, J. M. Clemente-Juan, B. Donnadieu, V. Nastopoulos, A. Mari, Y. Coppel, J. P. Tuchagues and S. P. Perlepes, Chem. – Eur. J., 2008, 14, 2514–2526 CrossRef CAS PubMed; (b) A. K. Boudalis, B. Donnadieu, V. Nastopoulos, J. M. Clemente-Juan, A. Mari, Y. Sanakis, J.-P. Tuchagues and S. P. Perlepes, Angew. Chem., Int. Ed., 2004, 43, 2266–2270 CrossRef CAS PubMed.
- G. S. Papaefstathiou, S. P. Perlepes, A. Escuer, R. Vicente, M. Font-Bardia and X. Solans, Angew. Chem., Int. Ed., 2001, 40, 884–886 CrossRef CAS PubMed.
- G. S. Papaefstathiou, A. Escuer, R. Vicente, M. Font-Bardia, X. Solans and S. P. Perlepes, Chem. Commun., 2001, 2414–2415 RSC.
- K. Meyer, E. Bill, B. Mienert, T. Weyhermüller and K. Wieghardt, J. Am. Chem. Soc., 1999, 121, 4859–4876 CrossRef CAS.
- J. Torres-Alacan, U. Das, A. C. Filippou and P. Vöhringer, Angew. Chem., Int. Ed., 2013, 52, 12833–12837 CrossRef CAS PubMed.
- J. Torres-Alacan, J. Lindner and P. Vöhringer, ChemPhysChem, 2015, 16, 2289–2293 CrossRef CAS PubMed.
- W. D. Wagner and K. Nakamoto, J. Am. Chem. Soc., 1989, 111, 1590–1598 CrossRef CAS.
- J. Chen and W. R. Browne, Coord. Chem. Rev., 2018, 374, 15–35 CrossRef CAS.
- J. Akhavan, Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2004, vol. 10, Explosives and Propellants, pp. 719–744, and references therein Search PubMed.
- (a) T. M. Klapötke, in High Energy Density Materials, ed. T. M. Klapötke, Springer, Berlin, Heidelberg, 2007, p. 85 CrossRef; (b) R. D. Chapman, in High Energy Density Materials, ed. T. M. Klapötke, Springer, Berlin, Heidelberg, 2007, p. 123 Search PubMed.
- T. M. Klapötke, Chemistry of High-Energy Materials, De Gruyter, Berlin, Boston, 4th edn, 2017 Search PubMed.
- D. M. Badgujar, M. B. Talawar, S. N. Asthana and P. P. Mahulikar, J. Hazard. Mater., 2008, 151, 289–305 CrossRef CAS PubMed.
- J. Köhler and J. Meyer, Explosivstoffe, Wiley-VCH, D-Weinheim, 9th edn, 1998 Search PubMed.
- (a) M. Talawar, R. Sivabalan, T. Mukundan, H. Muthurajan, A. Sikder, B. Gandhe and A. S. Rao, J. Hazard. Mater., 2009, 161, 589–607 CrossRef CAS PubMed; (b) M. A. Petrie, J. A. Sheehy, J. A. Boatz, G. Rasul, G. K. Surya Prakash, G. A. Olah and K. O. Christe, J. Am. Chem. Soc., 1997, 119, 8802–8808 CrossRef CAS.
- U. R. Nair, S. N. Asthana, A. S. Rao and B. R. Gandhe, Def. Sci. J., 2010, 60, 137–151 CrossRef CAS.
- M. W. Wemple, D. M. Adams, K. S. Hagen, K. Folting, D. N. Hendrickson and G. Christou, J. Chem. Soc., Chem. Commun., 1995, 1591–1593 RSC.
- S. M. J. Aubin, M. W. Wemple, D. M. Adams, H.-L. Tsai, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1996, 118, 7746–7754 CrossRef CAS.
- R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011–1026 RSC.
- Th. C. Stamatatos, A. Vinslava, K. A. Abboud and G. Christou, Chem. Commun., 2009, 2839–2841 RSC.
- (a) O. Kahn, Molecular Magnetism, VCH Publishers, New York, 1993 Search PubMed; (b) T. C. Stamatatos and G. Christou, Philos. Trans. R. Soc., A, 2008, 366, 113–125 CrossRef CAS PubMed.
- D. I. Alexandropoulos, A. Fournet, L. Cunha-Silva, G. Christou and Th. C. Stamatatos, Inorg. Chem., 2016, 55, 12118–12121 CrossRef CAS PubMed.
- D. I. Alexandropoulos, L. Cunha-Silva, A. Escuer and Th. C. Stamatatos, Chem. – Eur. J., 2014, 20, 13860–13864 CrossRef CAS PubMed.
- D. I. Alexandropoulos, K. R. Vignesh, Th. C. Stamatatos and K. R. Dunbar, Chem. Sci., 2018 10.1039/C8SC04384A.
- H. W. L. Fraser, G. S. Nichol, A. Baldansuren, E. J. L. McInnes and E. K. Brechin, Dalton Trans., 2018, 47, 15530–15537 RSC.
- For representative papers, see: (a) J. Ribas, A. Escuer, M. Monfort, R. Vicente, R. Cortes, L. Lezama and T. Rojo, Coord. Chem. Rev., 1999, 193–195, 1027–1068 CrossRef CAS; (b) C. Adhikary and S. Koner, Coord. Chem. Rev., 2010, 254, 2933–2958 CrossRef CAS; (c) E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Am. Chem. Soc., 1998, 120, 11122–11129 CrossRef CAS; (d) A. Escuer, R. Vicente, M. A. S. Goher and F. A. Mautner, Inorg. Chem., 1996, 35, 6386–6391 CrossRef CAS PubMed.
- S. M. J. Aubin, Z. Sun, H. J. Eppley, E. M. Rumberger, I. A. Guzei, K. Folting, P. K. Gantzel, A. L. Rheingold, G. Christou and D. N. Hendrickson, Inorg. Chem., 2001, 40, 2127–2146 CrossRef CAS PubMed.
- A. K. Boudalis, Y. Sanakis, J. M. Clemente-Juan, A. Mari and J. P. Tuchagues, Eur. J. Inorg. Chem., 2007, 2409–2415 CrossRef CAS.
- H. Oshio, N. Hoshino, T. Ito and M. Nakano, J. Am. Chem. Soc., 2004, 126, 8805–8812 CrossRef CAS PubMed.
- D. I. Alexandropoulos, K. R. Dunbar and T. C. Stamatatos, manuscript in preparation.
- (a) Y.-Z. Zhang, W. Wernsdorfer, F. Pan, Z.-M. Wang and S. Gao, Chem. Commun., 2006, 3302–3304 RSC; (b) Y.-L. Zhou, M.-H. Zeng, L.-Q. Wei, B.-W. Li and M. Kurmoo, Chem. Mater., 2010, 22, 4295–4303 CrossRef CAS.
- For a tutorial review on Co SMMs, see: M. Murrie, Chem. Soc. Rev., 2010, 39, 1986–1995 RSC.
- J. Krause, L. M. Carrella, E. Rentschler and T. C. Stamatatos, manuscript in preparation.
- J. Krause, D. I. Alexandropoulos, L. M. Carrella, E. Rentschler and Th. C. Stamatatos, Chem. Commun., 2018, 54, 12499–12502 RSC.
- Y.-K. Deng, H.-F. Su, J.-H. Xu, W.-G. Wang, M. Kurmoo, S.-C. Lin, Y.-Z. Tan, J. Jia, D. Sun and L.-S. Zheng, J. Am. Chem. Soc., 2016, 138, 1328–1334 CrossRef CAS PubMed.
- A. Hosoi, Y. Yukawa, S. Igarashi, S. J. Teat, O. Roubeau, M. Evangelisti, E. Cremades, E. Ruiz, L. A. Barrios and G. Aromí, Chem. – Eur. J., 2011, 17, 8264–8268 CrossRef CAS PubMed.
- M. Evangelisti, Molecule-Based Magnetic Coolers: Measurement, Design and Application, in Molecular Magnets: Physics and Applications, ed. J. Bartolomé, F. Luis, J. F. Fernández, Springer, Verlag, Berlin Heidelberg, 2014, pp. 365–387 Search PubMed.
- D. I. Alexandropoulos, L. Cunha-Silva, A. Escuer and T. C. Stamatatos, manuscript in preparation.
- For mononuclear 3d-SMMs exhibiting two relaxation processes, see: (a) J. M. Zadrozny and J. R. Long, J. Am. Chem. Soc., 2011, 133, 20732–20734 CrossRef CAS PubMed; (b) F. Habib, O. R. Luca, V. Vieru, M. Shiddiq, I. Korobkov, S. I. Gorelsky, M. K. Takase, L. F. Chibotaru, S. Hill, R. H. Crabtree and M. Murugesu, Angew. Chem., Int. Ed., 2013, 52, 11290–11293 CrossRef CAS PubMed; (c) F. Habib, I. Korobkov and M. Murugesu, Dalton Trans., 2015, 44, 6368–6373 RSC; (d) J. Miklovič, D. Valigura, R. Boča and J. Titiš, Dalton Trans., 2015, 44, 12484–12487 RSC; (e) J. Li, Y. Han, F. Cao, R.-M. Wei, Y.-Q. Zhang and Y. Song, Dalton Trans., 2016, 45, 9279–9284 RSC; (f) L. T. A. Ho and L. F. Chibotaru, Phys. Rev. B, 2016, 94, 104422 CrossRef.
- K. S. Pedersen, M. Sigrist, M. A. Sørensen, A.-L. Barra, T. Weyhermüller, S. Piligkos, C. A. Thuesen, M. G. Vinum, H. Mutka, H. Weihe, R. Clérac and J. Bendix, Angew. Chem., Int. Ed., 2014, 53, 1351–1354 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |