
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlkynylation of graphene via the Sonogashira C–C cross-coupling reaction on fluorographene†
Demetrios D.
Chronopoulos‡
 a,
Miroslav
Medved’‡
a,
Piotr
Błoński
a,
Zdeněk
Nováček
b,
Petr
Jakubec
a,
Ondřej
Tomanec
a,
Aristides
Bakandritsos
a,
Veronika
Novotná
b,
Radek
Zbořil
a and
Michal
Otyepka
*a
a,
Miroslav
Medved’‡
a,
Piotr
Błoński
a,
Zdeněk
Nováček
b,
Petr
Jakubec
a,
Ondřej
Tomanec
a,
Aristides
Bakandritsos
a,
Veronika
Novotná
b,
Radek
Zbořil
a and
Michal
Otyepka
*a
aRegional Centre for Advanced Technologies and Materials, Department of Physical Chemistry, Faculty of Science, Palacký University Olomouc, 17. listopadu 1192/12, 771 46 Olomouc, Czech Republic. E-mail: michal.otyepka@upol.cz
bNenoVision s.r.o., Purkyňova 649/127, 612 00 Brno, Czech Republic
First published on 7th January 2019
Abstract
We report successful grafting of alkynyl groups onto graphene via the Sonogashira reaction between fluorographene and terminal alkynes. Theoretical calculations revealed that fluorographene can efficiently bind and oxidize the palladium catalyst on electrophilic sites activated by fluorine atoms. This paves the way towards conductive and mechanically robust 3D covalent networks.
Graphene is considered a wonder material of the 21st century owing to its unique physical properties that can be exploited in countless applications.1–4 However, its zero band-gap,5 low chemical reactivity and poor dispersibility in polar media limit its applications, e.g., in biodisciplines.3 Hence, there is currently much interest in the preparation of graphene derivatives with tunable band-gap, controllable structural arrangements and tailored physicochemical properties. These features can be imprinted on graphene by covalent functionalization.6,7 Research into the scalable and controllable chemistry of fluorographene (FG) has matured to provide efficient synthetic approaches for preparing numerous graphene derivatives with tailored features.8–10 Cyanographene, graphene acid, and alkylated, alkenylated and arylated graphenes are prototypical examples of graphene derivatives successfully prepared from FG.11–13
Despite these efforts, grafting of rigid functional groups, to establish thermal and/or electrical conductivity perpendicular to a graphene surface, remains a challenge. Graphene equipped with such functional groups is a prerequisite for the synthesis of conductive and, at the same time, mechanically robust three-dimensional meshes, i.e., conductive graphene-based covalent networks. Alkynyl moieties represent ideal chemical groups fulfilling the abovementioned requirements. However, to date, published attempts for grafting an alkynyl group onto graphene have not been successful. For instance, Grignard compounds have been shown to be effective reagents for FG, providing alkylated, arylated and alkenylated graphenes.12,14 However, grafting of an ethynyl group onto FG was not possible using the Grignard reaction.12
Organometallic chemistry offers a variety of palladium-catalyzed C–C cross-coupling reactions between organic electrophiles and organometallic nucleophiles. In particular, in the Sonogashira reaction,15 coupling occurs between an aryl halide, vinyl halide or triflate and a terminal acetylene derivative in the presence of a palladium catalyst, a copper co-catalyst and an amine base. Based on the same concept, cross-coupling between alkyl halides and terminal alkynes has been achieved.16,17 The Sonogashira reaction has also been applied to modified graphene derivatives containing halogens.18,19 The drawbacks of this approach are the required pre-functionalization of graphene and the low amount of halogens. Moreover, in all previous cases, the triple bond was not attached directly onto graphene. All the above observations motivated us to consider FG as a potential platform for C–C cross-coupling via the Sonogashira reaction.
However, the C–F bond is the strongest single covalent bond to carbon.20 Although it can be activated by transition metal catalysts, participating in many organic reactions,21 fluorine compounds are not often used for Sonogashira coupling. To the best of our knowledge, there is only one report where alkynylation of polyfluorinated nitrobenzene compounds was carried out using palladium as the catalyst.22 The limited utilization of fluorine derivatives in the Sonogashira reaction is apparently related to the oxidation step requiring breaking of the C–X bond, which is energetically demanding for ordinary fluorine compounds. On the other hand, tertiary C–F bonds (i.e., bonds where F atoms are bound to tertiary carbons) in highly fluorinated polycyclic hydrocarbons are more susceptible to the reaction.23 Moreover, in the case of partially fluorinated graphene (pFG), carbon atoms in the vicinity of C–F bonds can act as efficient electrophilic sites that can assist in the C–F bond breaking by binding as well as oxidizing the Pd catalyst. Therefore, we considered the Sonogashira reaction using FG as a promising halide substrate for grafting alkyne moieties onto graphene.
Here, we report the successful synthesis of three alkynylated graphene derivatives via the palladium-catalyzed C–C cross-coupling Sonogashira reaction of FG with terminal alkynes, phenylacetylene (Pha), 4-ethynyl aniline (Eta), and 5-ethynylpyrimidine (Etp) (Fig. 1). Bis(triphenylphosphine) palladium(II) dichloride [Pd(Ph3P)2Cl2], copper iodide (CuI) and triethylamine (Et3N) were employed as catalyst, co-catalyst and base, respectively. All the reactions were performed in dry N,N-dimethylformamide (DMF) under oxygen-free conditions. The reaction yields of the prepared phenylacetylene graphene (PhaG), 4-ethynylaniline graphene (EtaG) and ethynylpyrimidine graphene (EtpG) were 45, 42 and 47%, respectively. The prepared graphene derivatives were dispersible in nonpolar solvents (toluene). The role of each of the reagents and catalysts participating in the Sonogashira reaction was investigated through many control experiments (see Section 1.3 in the ESI†) and insights into the reaction mechanism were gained via theoretical calculations.
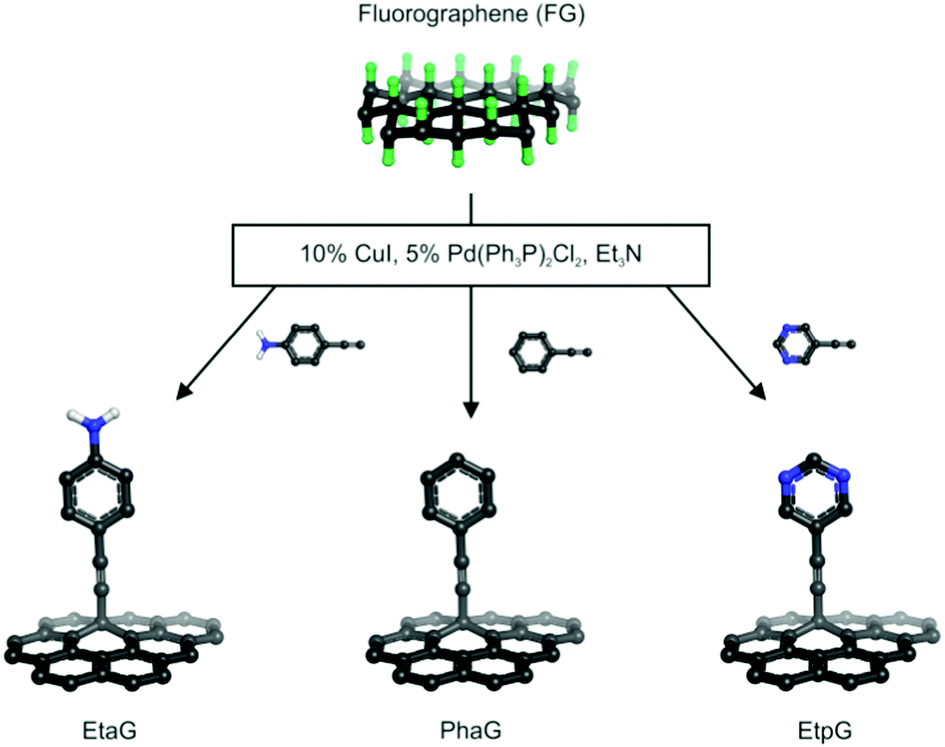 | ||
| Fig. 1 Overview of the Sonogashira reactions carried out on fluorographene. | ||
Successful grafting of alkynyl groups onto a graphene surface was confirmed by Fourier-transform infrared (FT-IR) spectroscopy. FT-IR spectra of the prepared alkynylated graphene derivatives (Fig. 2) showed that after the reaction, there was a lower peak at around 1200 cm−1, ascribed to the C–F bonds, compared to the corresponding peak of the starting material graphite fluoride (GF). Moreover, two characteristic bands appeared at around 1580 and 1450 cm−1, verifying the formation of conjugated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds due to reductive defluorination.24 Triple bonds in FT-IR spectra are usually difficult to detect because their corresponding intensity in disubstituted alkynes is very low and often missed. However, in the case of EtaG, a weak peak appeared at 2185 cm−1. Though less pronounced in the spectra of PhaG and EtpG, a weak band ascribable to a triple bond was also observed at 2024 and 1957 cm−1, respectively. As PhaG and EtpG contain a mono-substituted aromatic ring, the corresponding band was observed at 753 and 716 cm−1, respectively, whereas para di-substitution of the benzene ring (Bz-R) of EtaG gave rise to a peak at 828 cm−1. Finally, N–H vibrations occurred at 3358 cm−1.
C double bonds due to reductive defluorination.24 Triple bonds in FT-IR spectra are usually difficult to detect because their corresponding intensity in disubstituted alkynes is very low and often missed. However, in the case of EtaG, a weak peak appeared at 2185 cm−1. Though less pronounced in the spectra of PhaG and EtpG, a weak band ascribable to a triple bond was also observed at 2024 and 1957 cm−1, respectively. As PhaG and EtpG contain a mono-substituted aromatic ring, the corresponding band was observed at 753 and 716 cm−1, respectively, whereas para di-substitution of the benzene ring (Bz-R) of EtaG gave rise to a peak at 828 cm−1. Finally, N–H vibrations occurred at 3358 cm−1.
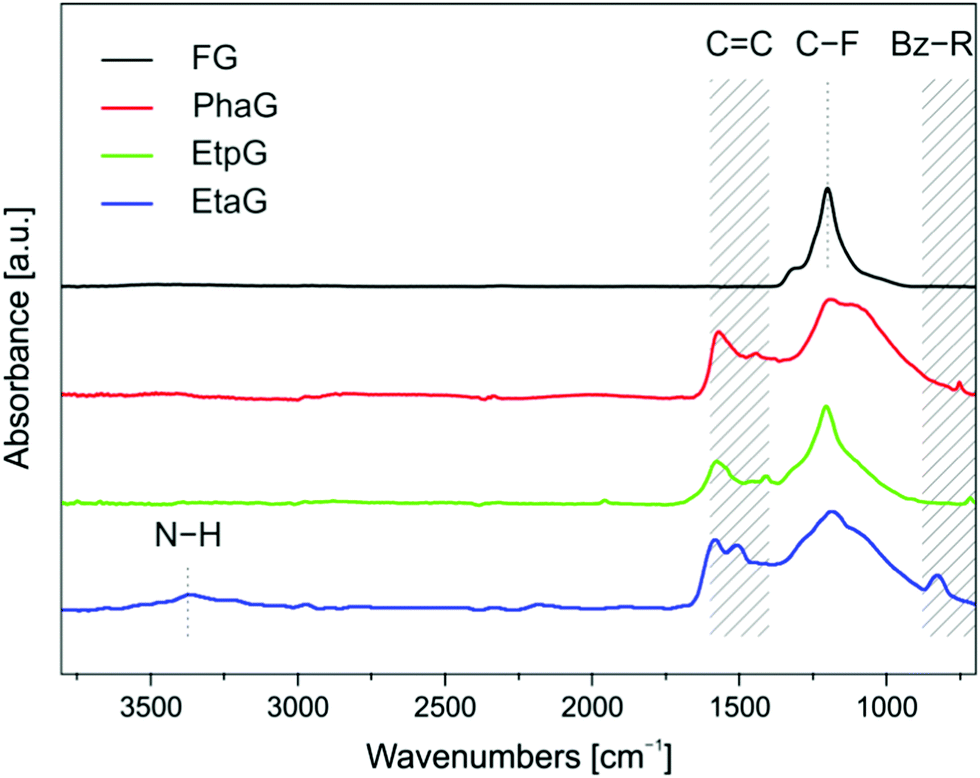 | ||
| Fig. 2 FT-IR spectra of graphene derivatives (PhaG, EtaG and EtpG) and pristine FG. | ||
According to X-ray photoelectron spectroscopy (XPS), all the graphene derivatives exhibited a significant reduction of fluorine atoms with respect to GF (Fig. S6–S10 and Table S1, ESI†). The C/F atomic ratios were 6.6, 23.2, and 3.34 for PhaG, EtaG and EtpG, respectively. As already mentioned, fluorine atoms were also removed at a low rate during the control experiment with FG, the catalysts and internal alkyne (1.3 for C/F). However, the significantly higher elimination of fluorine observed during the reaction with terminal alkynes indicated that defluorination was associated with successful conjugation of the terminal alkynes onto FG. Moreover, when nitrogen-containing alkynes were attached onto the formed graphene lattice, the amount of nitrogen detected was 7.6 and 5.4% for EtaG and EtpG, respectively, again confirming successful covalent functionalization (Table S1, ESI†).
Raman spectroscopy provided further evidence of the reduction of FG and its conversion to the corresponding graphene derivatives. Whereas pristine GF is not Raman active,25 the Raman spectra of the graphene derivatives showed the formation of D and G bands at characteristic values of 1320 and 1600 cm−1, respectively, indicating the formation of aromatic rings due to the partial defluorination (G-band) and formation of modified graphene derivatives (D-band). The intensity ratios (ID/IG) of the PhaG, EtaG and EtpG derivatives were 1.29, 1.11 and 1.15, respectively (Fig. S11–S13, ESI†).
Thermogravimetric analysis (TGA) provided important information about the thermal stability of the graphene derivatives. FG is thermally stable up to 400 °C and its decomposition occurs between 450 and 650 °C, losing 75% of its mass.25 Decomposition of the alkynylated graphene derivatives started at 180 °C and PhaG, EtaG and EtpG lost 21.7%, 25.4% and 38.9% of their weights, respectively, in the temperature range 180–580 °C (Fig. S14, ESI†). EtpG showed the highest weight loss, most likely because its graphenic structure was less stable due to the high percentage of fluorine atoms remaining after the modification.
Details on the morphologies of the graphene derivatives were obtained by high resolution transmission electron microscopy (HR-TEM), atomic force microscopy (AFM) and energy dispersive spectroscopy (EDS) elemental analysis. All the prepared materials consisted of a very low number of graphene sheets and showed similar morphological characteristics (Fig. 3A and B and Fig. S15–S17, ESI†). The thickness of EtpG was found to be about 5.0 nm from AFM measurements (Fig. 3G and H), indicating that the number of layers did not exceed three (one sheet of EtpG was predicted to be ca. 1.7 nm according to theoretical modeling; vide infra). EDS elemental mapping of EtpG showed the uniform presence of N and F on the graphene layer, verifying the homogeneous reductive defluorination of FG and homogeneous distribution of attached alkyne units over the formed graphene lattice (Fig. 3C–F).
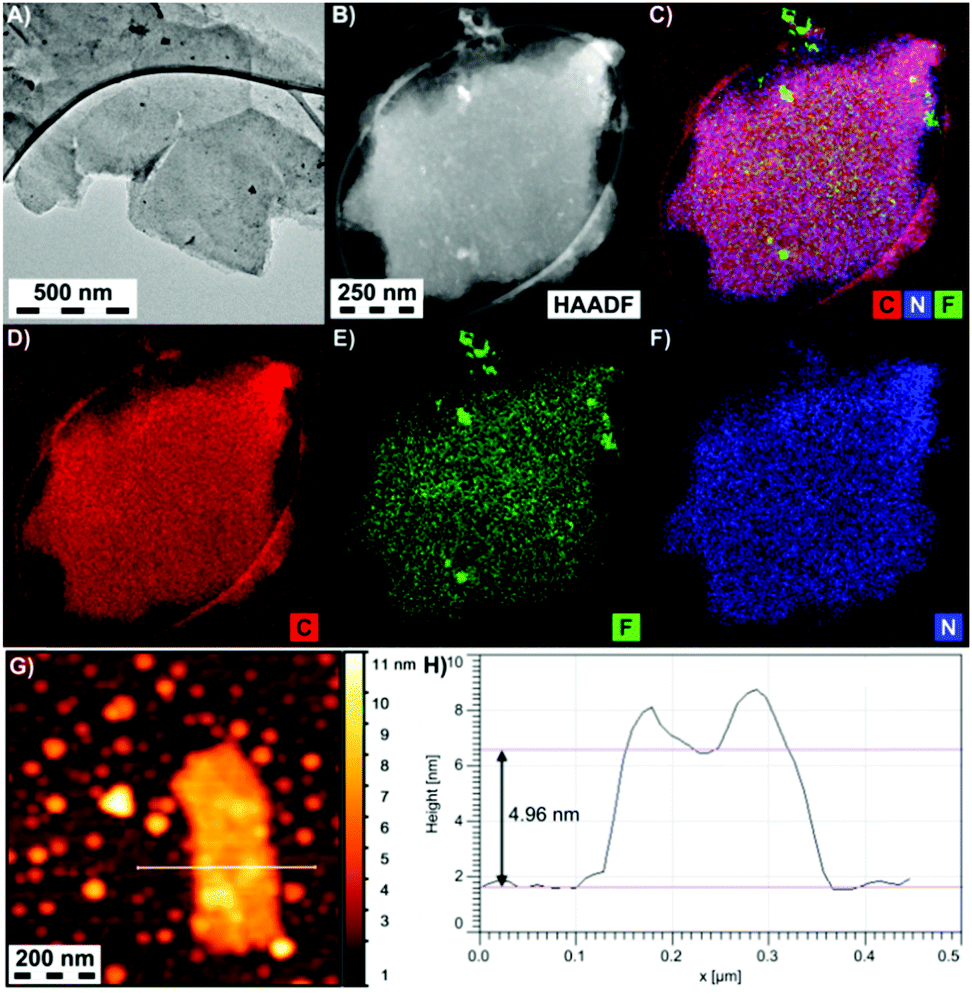 | ||
| Fig. 3 (A) HR-TEM and (B) dark field images of EtpG, (C–F) carbon, fluorine and nitrogen EDS maps of EtpG flakes, and (G and H) AFM image and the height profile of an EtpG flake. | ||
To rationalize the susceptibility of FG for the Sonogashira reaction, we explored the major steps of the reaction mechanism using density functional theory (DFT) calculations (see also Section 2.2 in the ESI†). Based on previous studies26–28 and our computational analysis, we adapted a generally accepted consensus mechanism for partially fluorinated graphene (pFG) acting as a substrate (Fig. 4A). Since pFG can provide a wide range of structural motifs for the reaction, various binding modes for the oxidative addition (step 1) were investigated (Fig. S24 and Table S4, ESI†). It was found that the PdL2 catalyst bound preferentially to carbon atoms in the vicinity of highly fluorinated regions. The most stable complex, with a binding energy of −18 kcal mol−1, was formed between PdL2 and a FG structure containing a fluorine vacancy. Such defects in FG were recently identified as highly reactive electrophilic sites.29 Solvent effects had only a minor effect on the binding characteristics of neutral FG⋯PdL2 complexes (Table S5, ESI†). In contrast to the usual organic substrates for this type of reaction, for which oxidative addition is an energetically demanding step due to the cleavage of the C–halogen bond,27 in pFG, the catalyst bound with a low activation barrier (∼11 kcal mol−1; Fig. S25, ESI†). The binding was accompanied by oxidation of the catalyst (Fig. S26, ESI†). Electron transfer from PdL2 to pFG played a crucial role in further steps of the mechanism as it promoted release of F− from pFG. Whereas the formation of the pFG–PdL2F complex (step 2) was energetically unfavorable (Fig. S27, ESI†), stabilization of the released fluoride anions in polar solvents (Fig. S29, ESI†) facilitated their pairing with Cu+ cations (step 3) and activation of the FG⋯PdL2 structure toward further attack by an alkynyl group (Fig. S28, ESI†). Although the formation of the pFG–PdL2(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–R2) complex (step 5) was also energetically allowed, we found that the alkynyl group preferentially bound directly onto pFG (step 4; see also Fig. S30, ESI†). Finally, reductive elimination leading to a product (step 6) required a relatively small energy (ca. 15–25 kcal mol−1). To sum up, the key features of pFG making it susceptible for the Sonogashira reaction were its ability to efficiently bind and oxidize the Pd catalyst on electrophilic sites activated by the presence of neighboring fluorine atoms, easing the oxidative addition, which is normally an energetically very demanding step for molecular fluorinated organic substrates.
C–R2) complex (step 5) was also energetically allowed, we found that the alkynyl group preferentially bound directly onto pFG (step 4; see also Fig. S30, ESI†). Finally, reductive elimination leading to a product (step 6) required a relatively small energy (ca. 15–25 kcal mol−1). To sum up, the key features of pFG making it susceptible for the Sonogashira reaction were its ability to efficiently bind and oxidize the Pd catalyst on electrophilic sites activated by the presence of neighboring fluorine atoms, easing the oxidative addition, which is normally an energetically very demanding step for molecular fluorinated organic substrates.
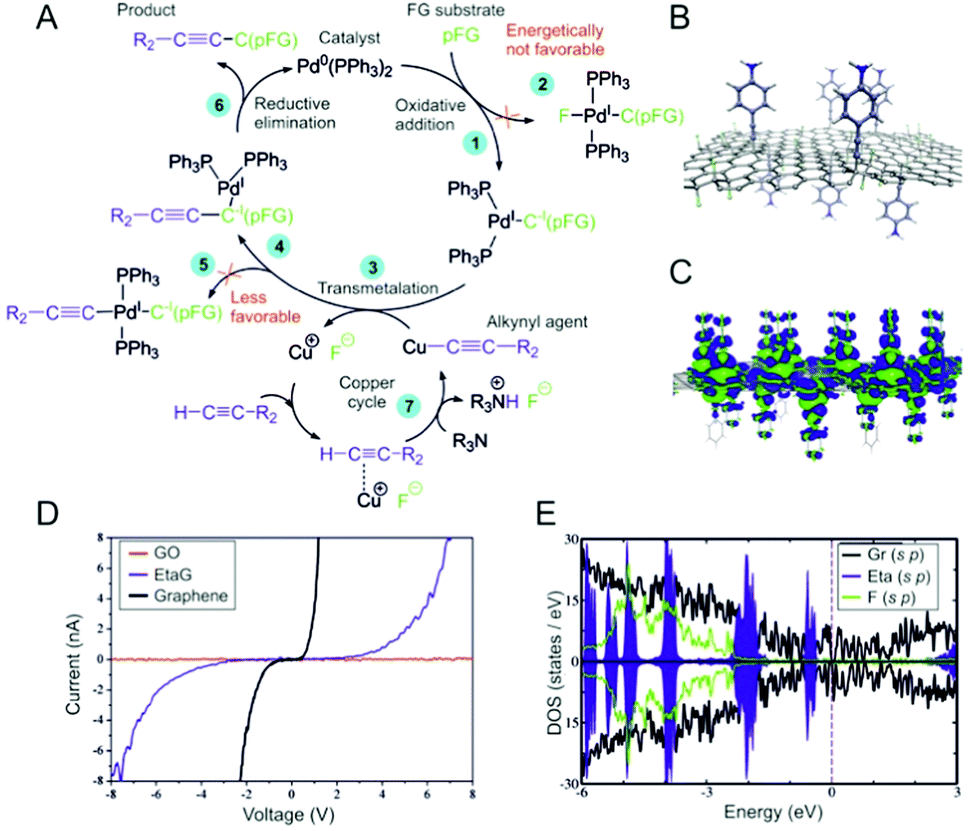 | ||
| Fig. 4 (A) Consensus mechanism of the Sonogashira cross-coupling reaction adapted for pFG. (B) Structural model of grafting Eta onto partially fluorinated graphene EtaG applied in the computations. (C) Electron density difference plot for PhaG (the contour isovalue is 10−3 e Å−3). (D) I/V spectroscopic curves of graphene (black line), EtaG (blue line) and GO (orange line). (E) DOS of the Eta-functionalized graphene; the energies are zeroed to the Fermi level; positive/negative DOS correspond to spin-up/spin-down electrons, respectively. | ||
The conductivity of EtaG was examined via correlative probe and electron microscopy (CPEM) (Fig. S18, ESI†). We chose the EtaG derivative for measurement of the I/V curve because it contained the least amount of fluorine atoms. Thus, its structure was closer to a “pure” alkynylated graphene derivative than the other derivatives and its conductivity had less influence from the fluorines. According to the measurements, the EtaG derivative was conductive, less compared to graphene but more compared to graphene oxide (GO) (Fig. 4D and the ESI† section for details of the measurement). Regarding EtpG, electrochemical impedance spectroscopy (EIS) also corroborated the higher conductivities of the alkynylated graphene derivatives with respect to GO (Fig. S19B, ESI†) and galvanostatic charge/discharge (GCD) curves presented promising results in the field of energy storage (Fig S19A, ESI†).
The theoretical electronic structures of the graphene derivatives presented as density of states (DOS) plots (Fig. 4E and Fig. S20, ESI†) revealed a significant reduction of the band-gap with respect to FG11 and GO.30 The electronic gaps of both a bare graphene layer and a partially fluorinated graphene sheet grafted with the functional groups did not exceed several tens of meV. A larger electronic gap in functionalized FG compared to functionalized graphene can be attributed to a greater number of carbon atoms changing their sp2 hybrid states to sp3 by the covalent functionalization, which may compromise the conductivity of the former material. Nonetheless, a pronounced density of states near the Fermi level (EF) conformed to the measured notable conductivity of the EtaG derivative. Further, Bader charge analysis indicated that up to ∼0.17e was transferred from the graphene substrate to each of the functional groups. The electron density difference plot for PhaG (Fig. 4C) agreed with this finding and suggested that there was a charge flow from the sp3-carbon atom neighborhood to π-orbitals of the functional group.
The electronic-structure calculations also revealed an uneven distribution of the functional groups and –F on the bipartite lattice of graphene, which changed its sp2 hybrid states to sp3 by the covalent functionalization. This caused a split by exchange interaction of the spin-up and spin-down states, and thus magnetism in some structures. For the sake of completeness, it should be noted that similar magnetic ordering was recently reported to emerge in graphene covalently functionalized by –F and –OH groups.31
We have developed a novel and straightforward method for the preparation of alkynylated graphene derivatives. Starting from GF, a commercially available material, alkyne moieties were directly grafted onto a concurrently formed graphene lattice via the Sonogashira cross-coupling reaction. Our approach opens new avenues for the preparation of modified graphene derivatives via a single step directly onto graphene, e.g., for the synthesis of rigid and conductive 3D graphene scaffolds.
We acknowledge financial support from MEYS (LO1305, CZ.1.05/2.1.00/19.0377, CZ.02.1.01/0.0/0.0/16_019/0000754) and the ERC (683024 from the H2020). We thank M. Petr for XPS.
Conflicts of interest
There are no conflicts to declare.References
- K. S. Novoselov, V. I. Fal′ko, L. Colombo, P. R. Gellert, M. G. Schwab and K. Kim, Nature, 2012, 490, 192–200 CrossRef CAS PubMed.
- X. Yu, H. Cheng, M. Zhang, Y. Zhao, L. Qu and G. Shi, Nat. Rev. Mater., 2017, 2, 17046 CrossRef CAS.
- G. Reina, J. M. González-Domínguez, A. Criado, E. Vázquez, A. Bianco and M. Prato, Chem. Soc. Rev., 2017, 46, 4400–4416 RSC.
- M. F. El-Kady, Y. Shao and R. B. Kaner, Nat. Rev. Mater., 2016, 1, 16033 CrossRef CAS.
- G. Lu, K. Yu, Z. Wen and J. Chen, Nanoscale, 2013, 5, 1353–1368 RSC.
- G. Bottari, M. Á. Herranz, L. Wibmer, M. Volland, L. Rodríguez-Pérez, D. M. Guldi, A. Hirsch, N. Martín, F. D’Souza and T. Torres, Chem. Soc. Rev., 2017, 46, 4464–4500 RSC.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- D. D. Chronopoulos, A. Bakandritsos, M. Pykal, R. Zbořil and M. Otyepka, Appl. Mater. Today, 2017, 9, 60–70 CrossRef PubMed.
- D. Matochová, M. Medved’, A. Bakandritsos, T. Steklý, R. Zbořil and M. Otyepka, J. Phys. Chem. Lett., 2018, 9, 3580–3585 CrossRef PubMed.
- A. Bakandritsos, D. D. Chronopoulos, P. Jakubec, M. Pykal, K. Čépe, T. Steriotis, S. Kalytchuk, M. Petr, R. Zbořil and M. Otyepka, Adv. Funct. Mater., 2018, 28, 1801111 CrossRef.
- A. Bakandritsos, M. Pykal, P. Błoński, P. Jakubec, D. D. Chronopoulos, K. Poláková, V. Georgakilas, K. Čépe, O. Tomanec, V. Ranc, A. B. Bourlinos, R. Zbořil and M. Otyepka, ACS Nano, 2017, 11, 2982–2991 CrossRef CAS PubMed.
- D. D. Chronopoulos, A. Bakandritsos, P. Lazar, M. Pykal, K. Čépe, R. Zbořil and M. Otyepka, Chem. Mater., 2017, 29, 926–930 CrossRef CAS PubMed.
- W. Lai, J. Liu, L. Luo, X. Wang, T. He, K. Fan and X. Liu, Chem. Commun., 2018, 54, 10168–10171 RSC.
- V. Mazánek, A. Libánská, J. Šturala, D. Bouša, D. Sedmidubský, M. Pumera, Z. Janoušek, J. Plutnar and Z. Sofer, Chem. – Eur. J., 2017, 23, 1956–1964 CrossRef PubMed.
- R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- M. Eckhardt and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 13642–13643 CrossRef CAS PubMed.
- G. Altenhoff, S. Würtz and F. Glorius, Tetrahedron Lett., 2006, 47, 2925–2928 CrossRef CAS.
- X. Zhuang, F. Zhang, D. Wu, N. Forler, H. Liang, M. Wagner, D. Gehrig, M. R. Hansen, F. Laquai and X. Feng, Angew. Chem., Int. Ed., 2013, 52, 9668–9672 CrossRef CAS PubMed.
- R. Kumar, V. M. Suresh, T. K. Maji and C. N. R. Rao, Chem. Commun., 2014, 50, 2015–2017 RSC.
- D. O’Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed.
- M. R. Cargill, G. Sandford, P. Kilickiran and G. Nelles, Tetrahedron, 2013, 69, 512–516 CrossRef CAS.
- G. Sandford, Tetrahedron, 2003, 59, 437–454 CrossRef CAS.
- R. Zbořil, F. Karlický, A. B. Bourlinos, T. A. Steriotis, A. K. Stubos, V. Georgakilas, K. Šafářová, D. Jančík, C. Trapalis and M. Otyepka, Small, 2010, 6, 2885–2891 CrossRef PubMed.
- J. T. Robinson, J. S. Burgess, C. E. Junkermeier, S. C. Badescu, T. L. Reinecke, F. K. Perkins, M. K. Zalalutdniov, J. W. Baldwin, J. C. Culbertson, P. E. Sheehan and E. S. Snow, Nano Lett., 2010, 10, 3001–3005 CrossRef CAS PubMed.
- L. J. Gooßen, D. Koley, H. Hermann and W. Thiel, Chem. Commun., 2004, 2141–2143 RSC.
- S. Kozuch, C. Amatore, A. Jutand and S. Shaik, Organometallics, 2005, 24, 2319–2330 CrossRef CAS.
- L. Xue and Z. Lin, Chem. Soc. Rev., 2010, 39, 1692–1705 RSC.
- M. Medved’, G. Zoppellaro, J. Ugolotti, D. Matochová, P. Lazar, T. Pospíšil, A. Bakandritsos, J. Tuček, R. Zbořil and M. Otyepka, Nanoscale, 2018, 10, 4696–4707 RSC.
- M. Lundie, Ž. Šljivančanin and S. Tomić, J. Phys.: Conf. Ser., 2014, 526, 012003 CrossRef.
- J. Tuček, K. Holá, A. B. Bourlinos, P. Błoński, A. Bakandritsos, J. Ugolotti, M. Dubecký, F. Karlický, V. Ranc, K. Čépe, M. Otyepka and R. Zbořil, Nat. Commun., 2017, 8, 14525 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc08492k |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |