
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diverse outcomes of CO2 fixation using alkali metal amides including formation of a heterobimetallic lithium–sodium carbamato-anhydride via lithium–sodium bis-hexamethyldisilazide†
Richard M.
Gauld
a,
Alan R.
Kennedy
 a,
Ross
McLellan
a,
Jim
Barker
b,
Jacqueline
Reid
b and
Robert E.
Mulvey
*a
a,
Ross
McLellan
a,
Jim
Barker
b,
Jacqueline
Reid
b and
Robert E.
Mulvey
*a
aWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, Glasgow, G1 1XL, UK. E-mail: r.e.mulvey@strath.ac.uk
bInnospec Ltd, Innospec Manufacturing Park, Oil Sites Road, Ellesmere Port, Cheshire, CH65 4EY, UK
First published on 15th January 2019
Abstract
Fixation of CO2 by lithium amides derived from pyrrole and diisopropylamine generates a lithium carbamate polymer and dodecamer respectively. Moving to lithium–sodium hexamethyldisilazide produces a more complicated, intriguing reaction, where unusually the bimetallic composition is maintained in the product but its composition contains both carbamato and anhydride functionalities.
The activation and fixation of small molecules is a fertile field of research currently at the cutting edge of inorganic and organometallic chemistry.1–3 Along with fundamental interest, the goal of small molecule activation is to convert common small molecules such as N2, O2 and CO2 to higher value products, achieved through a variety of chemical means.1 For example, Arnold recently demonstrated small molecule activation of N2, CO and CO2 using a uranium based tris(aryloxide), giving a range of complexes such as [U(OAr)3]2(μ-N2) and the ynediolate complex of the form [U(OAr)3]2(μ-η1:η1-C2O2).4 Transition metals such as iron and manganese have been used to activate molecular oxygen, both in synthetic chemistry5 and in nature with the best known example in the transport of oxygen in blood of vertebrates by the iron protein haemoglobin.6,7
Carbon dioxide is a molecular Jekyll and Hyde, famous as it is the primary carbon source for life on earth; but infamous because of its role as a greenhouse gas. Attempts to capture and sequester CO2 are currently the focus of many scientific efforts worldwide.8 The stability of CO2 presents a significant challenge in terms of its activation due to the strong C–O double bonds present, exemplified by the high enthalpy value of −393.51 kJ mol−1.2,9 Nonetheless, there are many methods available in this regard. For example, nature uses enzymes that employ CO2 or its hydrated form to carry out a carboxylation reaction to form a new C–C bond.9 CO2 can also be utilised in the synthesis of polymers, as in the reaction with cyclohexane oxide catalysed by zinc crotonate.10
In spite of this widespread interest, few studies have looked at CO2 insertion into reactive alkali metal–nitrogen bonds,11 with much greater attention being paid to transition metals and group 14 elements. Such insertion reactions and concomitant formations of carbamate functionalities adds to the diversity of CO2 chemistry, which attracts widespread interest across capture technologies,12 industry and agriculture.13 The paucity of work carried out on alkali metal amides is perhaps surprising given the widespread utility of these amides in synthesis. Lithium diisopropylamide (LDA) is the most common utility amide, first utilised in 1950 in reactions with esters.14 The structure of LDA in the solid state was elucidated in 1991, revealing an eye-catching helical arrangement with near-linear N–Li–N–Li units,15 with a more recent study establishing an equilibrium of trimers and tetramers in toluene.16 Another class of compound that has received much synthetic interest are pyrroles. This is due to the prevalence of the five-membered ring in areas ranging from agricultural chemistry17 to the pharmaceutical industry.18
Herein we report the different outcomes of the insertion reactions of CO2 into common utility lithium amides, namely LDA and lithium pyrrolide. Extending our study to reactions between CO2 and the mixed lithium–sodium 1,1,1,3,3,3-hexamethyldisilazide (HMDS) system, THF·Li(μ-HMDS)2Na·2THF, reveals a remarkable carbamato-anhydride product, which retains both lithium and sodium in its polymeric chain structure.
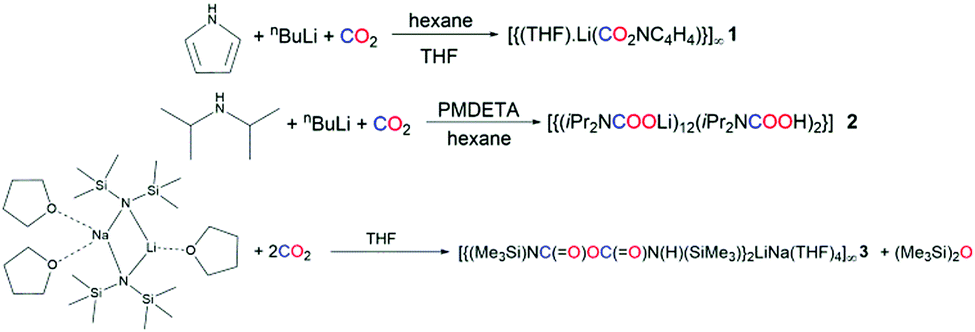
The lithium–nitrogen bonding in lithium pyrrolide has recently been elucidated, and suggests that the insertion of CO2 into lithium pyrrolide should be possible.19 To this end, pyrrole was reacted with nBuLi in hexane at 0 °C, then CO2 was bubbled through the resulting suspension for 30 minutes. Adding THF produced colourless crystals of the polymeric carbamate [{(THF)·Li(CO2NC4H4)}∞] (1) (Scheme 1), as determined by X-ray crystallography (Fig. 1).
 | ||
| Scheme 1 CO2 fixation reactions carried out in this work. | ||
 | ||
| Fig. 1 (Top) Threefold section of the polymeric structure of [{(THF)·Li(CO2NC4H4)}∞] 1; and (bottom) step-ladder arrangement of its 8-atom rings. H atoms are omitted for clarity and thermal ellipsoids are displayed at 40% probability level. Symmetry transformations used to generate equivalent atoms: −x, −y, −z. | ||
Carbamate 1 adopts a polymeric ladder of alternating, fused 4-membered (LiO)2 and 8-membered (LiO)4 rings, with a THF donor ligand completing the Li coordination. The structure contains only one Li environment, confirmed both in the solid and solution state (see ESI†).
This environment comprises four oxygen atoms in a distorted tetrahedron, three from the carbamate units and the fourth from THF. The mean Li–O–Li bond angle is 109°, ranging from 123.8(3)° to 90.5(2)° (τ4 = 0.81).20 The (LiO)2 ring forms a near perfect square, with internal angles of 90.5(2)° and 89.5(2)° at Li and O respectively. Least squares planes through the 4- and 8-membered rings make a dihedral angle of 138°. Though ladder motifs are well known in lithium amide chemistry21–25 to our knowledge this is the first such ladder motif found in lithium carbamate chemistry. The Li cation is μ3 with respect to the carbamate oxygens (Li–O range 1.870(5)–2.020(6) Å) and is effectively equidistant between the carbamate O atom and THF O atom [Li1–O2 and Li1–O3 distance 1.943(5) vs. 1.959(6) Å respectively]. The carbamate C–O distances are longer than those seen in the parent CO2 molecule8 and are intermediate between C–O single and double bonds. Robertson et al. probed the reaction products of CO2 insertion into LiTMP observing a similar distance for the C–O bond as seen in 1 [values of 1.255(2) and 1.261(2) Å].26 Snaith et al. also observed similar distances to those of 1 when studying CO2 insertion products of lithium diphenylamide, with values of 1.251(7)/1.256(7) Å for one carbamate unit and 1.264(7)/1.243(7) Å for the other.27 Due to the widespread nature of the five membered ring, it should come as no surprise to find that N–CO2 based structural motifs such as that seen in polymeric 1 are present in a variety of biologically active compounds, which have found use as potential cyclotoxic antitumor agents to antiretroviral therapies.28,29
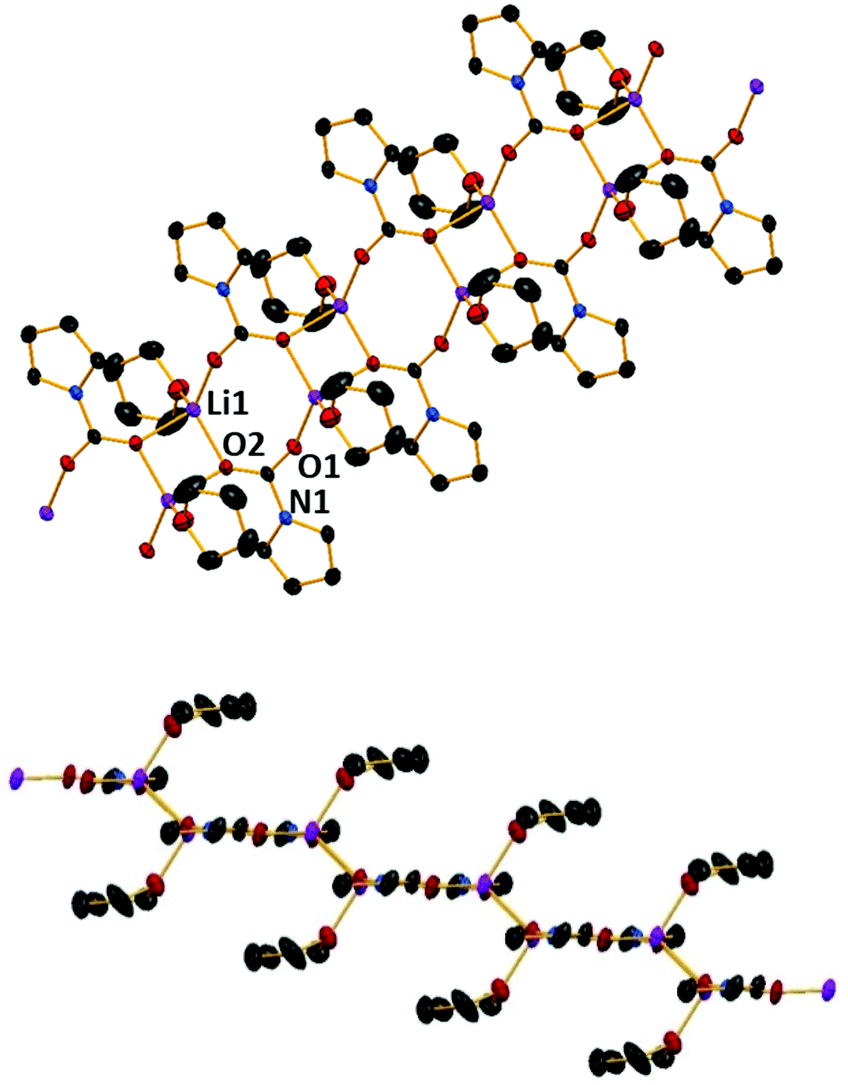
Next we turned to LDA. The reactive Li–N bond present would be ideal for use in CO2 activation presumably furnishing the corresponding lithium carbamate complex. To this end, diisopropylamine was deprotonated with nBuLi in hexane, N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) was introduced and CO2 was bubbled through this solution for 10 minutes. Cooling the resulting solution to −33 °C gave crystals identified as [{(iPr2NCOOLi)12(iPr2NCOOH)2}] (2) (Scheme 1 and Fig. 2) by X-ray crystallography.
 | ||
| Fig. 2 (LHS) Molecular structure of [{(iPr2NCOOLi)12(iPr2NCOOH)2}] (2). Hydrogen atoms are omitted and organic groups are shown as wire frame for clarity; (RHS) structure of its Li–O core with organic groups omitted for clarity. Thermal ellipsoids are displayed at 40% probability level. Symmetry transformations used to generate equivalent atoms: −x, −y, −z. | ||
The centrosymmetric structure of 2 is a discrete dodecanuclear cluster comprising 12 lithium diisopropylamido-carbamate units and two carbamic acids that occupy terminal positions. Note there is no PMDETA present in the structure, though subsequent experiments have shown that PMDETA addition is necessary in order to obtain crystals. The Li cations are bonded to three, four or five oxygen atoms, depending upon their position in the structure. Those bonded to three oxygen atoms (Li1) occupy a slightly distorted trigonal environment (average angle of 119°). The four coordinate Li centres are all in a distorted tetrahedral environment, with τ4 values ranging from 0.80–0.84 (Li2). There are two lithium centres in the molecule bonded to five oxygen atoms (Li3), both in a distorted square pyramidal molecular geometry (average bond angle of 94.7°).
The two carbamic acid groups present at the terminal positions of the Li–O core presumably result from adventitious hydrolysis, likely due to the CO2 source being dry ice. However, the reaction is reproducible and the CO2 is passed through a CaCl2 drying tube prior to addition. This is a relatively rare example of such a feature, with only a few examples of carbamic acid groups structurally characterised.30,31 In 2 the metallic core skeleton shows a distorted Li4O4 cubane that is linked to a perfect central Li2O2 planar square (all internal angles 90°) via a μ-(O–N) bond. The corner oxygen atoms of the cubane are connected to the remaining Li of the Li2O2 square by a carbamate in a κO;κO1 bridging mode. The C–O distances in the carbamate units within the cubane section of 2 are consistent with those seen in 1, with values generally in the range of 1.25–1.26 Å, again longer than in the parent CO2 molecule.8 The bridging carbamates have slightly different bond lengths, with one generally shorter than the other (1.235(4) vs. 1.275(5) and 1.267(3) vs. 1.288(3) Å), presumably due to the bridging nature.
The carbamic acid molecules have bond lengths that would be expected for a carboxylic acid derived species, with a C–O double bond of 1.227(3) Å and a C–OH single bond of 1.321(4) Å, though the OH group is not visible in either IR or NMR spectra, perhaps due to the lower number of such groups in comparison to the carbamate groups within the complex.
Diisopropylamide species have been used in CO2 activation before, notably by Sperrle who obtained a hafnium carbamate complex.32 Interestingly, the structure of this complex is simpler than the Li analogue here, having an 8-coordinate hafnium atom bound by four DA-derived carbamate units.32
We next attempted to expand fixation of CO2 into another major class of utility amide; namely those based on alkali metal HMDS compounds.33 Previous studies have shown M-HMDS species are able to activate CO2. Insertion into U-(HMDS)4 is followed by a series of silyl migration steps, furnishing an isocyanate complex.34 The presence of an isocyanate is also implicated by Kemp et al., who probed the insertion reactions of CO2 into group two complexes based on a HMDS core.35 We wanted to probe the effects of CO2 addition specifically on mixed alkali metal species, based on work carried out by Williard which disclosed the structures of these mixed alkali metal HMDS complexes to be dimeric.36 As such, these species would be primed for CO2 insertion in two distinct sites within the same molecule, between the Li–N bond and Na–N bond for example. However, to the best of our knowledge it is exceptionally rare to generate a mixed alkali metal product from a mixed alkali metal starting material.37,38
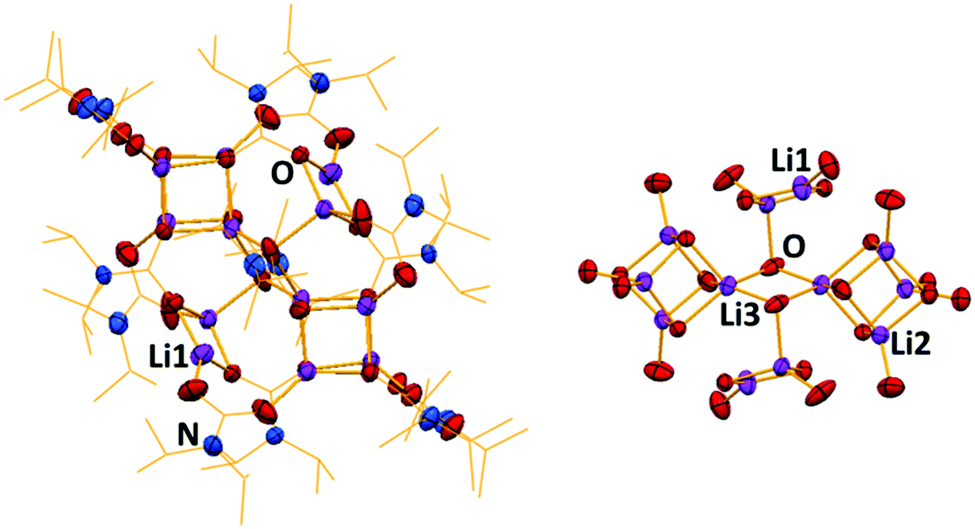
To this end, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 NaHMDS:LiHMDS mixture was dissolved in THF, and then CO2 was bubbled through it for 10 minutes at −78 °C. On warming to room temperature, a yellow solution formed. Cooling this solution to −33 °C deposited crystals, revealed by X-ray crystallography to be the unique lithium–sodium carbamato-anhydride polymer [{(Me3Si)NC(
:1 NaHMDS:LiHMDS mixture was dissolved in THF, and then CO2 was bubbled through it for 10 minutes at −78 °C. On warming to room temperature, a yellow solution formed. Cooling this solution to −33 °C deposited crystals, revealed by X-ray crystallography to be the unique lithium–sodium carbamato-anhydride polymer [{(Me3Si)NC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OC(O)N(H)(SiMe3)}2LiNa(THF)4]∞, (3) (Scheme 1 and Fig. 3). Repeated attempts confirmed the reproducibility of this reaction with yields spanning the range 45–51%. Disorder in THF and Me3Si groups in 3 rules out a discussion of its bond angles and lengths though its molecular connectivity is unambiguous. Interestingly, when LiHMDS and NaHMDS were reacted separately with CO2, only intractable gels were formed, implying the origin of 3 is synergistic.
O)OC(O)N(H)(SiMe3)}2LiNa(THF)4]∞, (3) (Scheme 1 and Fig. 3). Repeated attempts confirmed the reproducibility of this reaction with yields spanning the range 45–51%. Disorder in THF and Me3Si groups in 3 rules out a discussion of its bond angles and lengths though its molecular connectivity is unambiguous. Interestingly, when LiHMDS and NaHMDS were reacted separately with CO2, only intractable gels were formed, implying the origin of 3 is synergistic.
 | ||
| Fig. 3 Section of the polymeric structure of alkali metal carbamato-anhydride [{(Me3Si)NC(O)OC(O)N(H)(SiMe3)}2LiNa(THF)4] (3). H atoms and THF molecules are omitted for clarity; the bottom image also has non-carbamate carbon atoms omitted for clarity. Thermal ellipsoids are displayed at 40% probability level. Symmetry transformations used to generate equivalent atoms: −x, y, −z; 1/2+x, 1/2+y, z; 1/2−x, 1/2+y, −z. | ||
The structure of 3 exists as an infinite chain of silylamido-substituted anhydride units held together by alternating Li and Na cations. The Li cations are four-coordinate in a distorted tetrahedral environment (τ4 = 0.78), each coordinated to four oxygens from two chelating anhydride molecules. The Na cations are six-coordinate, bonded to two oxygen and two nitrogen atoms from two individual anhydride units. The nitrogen and oxygen atoms are in the backbone of the anhydride, thus directing polymer formation. The Na coordination sphere is completed by two THF ligands. Therefore the anhydride unit adopts a ditopic arrangement in terminally binding to Na through one O centre and chelating the Li through two oxygen centres on the opposite face.
Given the unexpected nature of 3, a literature search was done which found no evidence for crystallographically characterised alkali metal anhydrides. However, CO2 has previously been used in the synthesis of an anhydride derived compound, namely hexahydro-4,7-dimethyl-1-oxatetrazonine-2,9-dione.39 The synthesis involved the addition of CO2 to either a mixture of the parent dihydrizine and HN(SiMe3)2 or directly reacting CO2 with the corresponding disilane.39
Based on the stoichiometry of 3, it appears that two trimethylsilyl units and one oxygen atom have been lost from the reaction, which also shows the formation of a new NH group. As for how compound 3 may have formed, Maron and Mazzanti investigated the conversion of CO2 to isocyanates at uranium centres, postulating that multiple N–Si bond cleavage reactions occurred, leading to the release of hexamethyldisiloxane.34 This could be a potential pathway in this reaction, explaining the loss of both the oxygen atom and the silylamido groups. The NH group present within the polymer backbone could have been formed either through adventitious hydrolysis or the presence of some HMDS(H) within the reaction. In any case, the anhydride can also be isolated in its hydrolysed form using H2O work-up, with the parent compound featuring two NH groups in the backbone.
In conclusion, we have demonstrated that common utility alkali metal amides can be utilised in CO2 insertion reactions, providing a route to the formation of alkali metal carbamate species. Through judicious choice of amide, different molecular architectures can be obtained, ranging from polymeric through to discrete species. A novel alkali metal anhydride species is also presented through the activation of CO2, revealing an exceptionally rare reaction where a mixed lithium–sodium precursor produces a mixed lithium–sodium product, itself a rare example of a heterobimetallic alkali metal polymer.
We thank the EPSRC (DTP award EP/N509760/1) and Innospec Ltd for funding the studentship of RMG. We also thank both Craig Irving for help with NMR aspects and Dr Charles O’Hara for helpful discussions. Data used within this publication can be accessed at https://dx.doi.org/10.15129/1ae0a46f-614f-4994-9d61-9473bab1807b.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Milani, G. Licini, E. Clot and M. Albrecht, Dalton Trans., 2016, 45, 14419–14420 RSC.
- W. B. Tolman, Activation of Small Molecules: Organometallic and Bioinorganic Perspectives, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006 Search PubMed.
- D. B. Dell'Amico, F. Calderazzo, L. Labella, F. Marchetti and G. Pampaloni, Chem. Rev., 2003, 103, 3857–3898 CrossRef PubMed.
- S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942–13958 CrossRef CAS PubMed.
- R. M. Wells, V. Tetens and T. Brittain, Nature, 1983, 306, 500–502 CrossRef CAS PubMed.
- C. Thomas and A. B. Lumb, Continuing Education in Anaesthesia Critical Care & Pain, 2012, 12, 251–256 Search PubMed.
- A. Paparo and J. Okuda, Coord. Chem. Rev., 2017, 334, 136–149 CrossRef CAS.
- M. Aresta, in Activation of Small Molecules: Organometallic and Bioinorganic Perspectives, ed. W. B. Tolman, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006, ch. 1, pp. 1–41 Search PubMed.
- D. J. Darensbourg and M. S. Zimmer, Macromolecules, 1999, 32, 2137–2140 CrossRef CAS.
- X. Yin and J. R. Moss, Coord. Chem. Rev., 1999, 181, 27–59 CrossRef CAS.
- L. J. Murphy, K. N. Robertson, R. A. Kemp, H. M. Tuononen and J. A. C. Clyburne, Chem. Commun., 2015, 51, 3942–3956 RSC.
- Q. Qin, Z. Guo and X. Wei, J. Mol. Struct., 2016, 1114, 156–160 CrossRef CAS.
- M. Hamell and R. Levine, J. Org. Chem., 1950, 162–168 CrossRef CAS.
- N. D. R. Barnett, R. E. Mulvey, W. Clegg and P. A. O'Neil, J. Am. Chem. Soc., 1991, 113, 8187–8188 CrossRef CAS.
- R. Neufeld, M. John and D. Stalke, Angew. Chem., Int. Ed., 2015, 54, 6994–6998 CrossRef CAS PubMed.
- Y. Zhao, C. Mao, Y. Li, P. Zhang, Z. Huang, F. Bi, R. Huang and Q. Wang, J. Agric. Food Chem., 2008, 56, 7326–7332 CrossRef CAS PubMed.
- G. La Regina, A. Coluccia, A. Brancale, F. Piscitelli, V. Gatti, G. Maga, A. Samuele, C. Pannecouque, D. Schols, J. Balzarini, E. Novellino and R. Silvestri, J. Med. Chem., 2011, 54, 1587–1598 CrossRef CAS PubMed.
- F. Engelhardt, C. Maaß, D. M. Andrada, R. Herbst-Irmer and D. Stalke, Chem. Sci., 2018, 9, 3111–3121 RSC.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- D. R. Armstrong, D. Barr, W. Clegg, S. M. Hodgson, R. E. Mulvey, D. Reed, R. Snaith and D. S. Wright, J. Am. Chem. Soc., 1989, 111, 4719–4727 CrossRef CAS.
- R. E. Mulvey, Chem. Soc. Rev., 1998, 27, 339–346 RSC.
- D. R. Armstrong, D. Barr, R. Snaith, W. Clegg, R. E. Mulvey, K. Wade and D. Reed, J. Chem. Soc., Dalton Trans., 1987, 1071–1081 RSC.
- H. Chen, R. A. Bartlett, H. V. Rasika Dias, M. M. Olmstead and P. P. Power, Inorg. Chem., 1991, 30, 2487–2494 CrossRef CAS.
- W. Clegg, S. H. Dale, D. V. Graham, R. W. Harrington, E. Hevia, L. M. Hogg, A. R. Kennedy and R. E. Mulvey, Chem. Commun., 2007, 1641–1643 RSC.
- A. R. Kennedy, R. E. Mulvey, D. E. Oliver and S. D. Robertson, Dalton Trans., 2010, 39, 6190–6197 RSC.
- S. C. Ball, M. G. Davidson, R. P. Davies, A. J. Edwards, I. Lopez-Solera, P. R. Raithby and R. Snaith, Angew. Chem., Int. Ed. Engl., 1995, 921–923 CrossRef CAS.
- C. Willemann, R. Gruenert, P. J. Bednarski and R. Troschuetz, Bioorg. Med. Chem., 2009, 17, 4406–4419 CrossRef CAS PubMed.
- X. Li and R. Vince, Bioorg. Med. Chem., 2006, 14, 2942–2955 CrossRef CAS.
- A. Berkessel, K. Roland, M. Schröder, J. M. Neudörfl and J. Lex, J. Org. Chem., 2006, 71, 9312–9318 CrossRef CAS PubMed.
- A. Esparza-Ruiz, C. Herrmann, J. Chen, B. O. Patrick, E. Polishchuk and C. Orvig, Inorg. Chim. Acta, 2012, 393, 276–283 CrossRef CAS.
- F. Calderazzo, S. Ianelli, G. Pampaloni, G. Pelizzi and M. Sperrle, J. Chem. Soc., Dalton Trans., 1991, 693–698 RSC.
- R. Neufeld, R. Michel, R. Herbst-Irmer, R. Schöne and D. Stalke, Chem. – Eur. J., 2016, 22, 12340–12346 CrossRef CAS PubMed.
- C. Camp, L. Chatelain, C. E. Kefalidis, J. Pécaut, L. Maron and M. Mazzanti, Chem. Commun., 2015, 51, 15454–15457 RSC.
- D. A. Dickie, K. B. Gislason and R. A. Kemp, Inorg. Chem., 2012, 51, 1162–1169 CrossRef CAS PubMed.
- P. G. Williard and M. A. Nichols, J. Am. Chem. Soc., 1991, 113, 9671–9673 CrossRef CAS.
- J. J. Morris, B. C. Noll and K. W. Henderson, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2477 CrossRef CAS.
- D. R. Armstrong, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2011, 17, 8820–8831 CrossRef CAS PubMed.
- A. D. Kirilin, L. O. Belova, A. V. Lega, A. S. Maksimov, S. V. Petrov and E. A. Chernyshev, Russ. J. Gen. Chem., 2005, 75, 1163–1164 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectroscopic and crystallographic details. CCDC 1873068–1873070. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc08308h |
| This journal is © The Royal Society of Chemistry 2019 |