
Pelargonidin-3-O-rutinoside as a novel α-glucosidase inhibitor for improving postprandial hyperglycemia†
Yang
Xu
a,
Lianghua
Xie
a,
Jiahong
Xie
a,
Yu
Liu
b and
Wei
Chen
 *a
*a
aDepartment of Food Science and Nutrition, National Engineering Laboratory of Intelligent Food Technology and Equipment, Zhejiang Key Laboratory for Agro-Food Processing, Fuli Institute of Food Science, Zhejiang University, Hangzhou 310058, China. E-mail: zjuchenwei@zju.edu.cn
bCollege of Life Science, Zhejiang University, Hangzhou 310058, China
First published on 25th October 2018
Abstract
A novel strategy for the isolation of anthocyanins was developed by a combination of high-speed countercurrent chromatography (HSCCC) and HPLC techniques. Using this strategy, a novel α-glucosidase inhibitor (pelargonidin-3-O-rutinoside, a natural anthocyanin) was isolated from strawberries. Structural determinants for inhibition of α-glucosidase by Pg3R and the SAR of natural anthocyanins were revealed in detail by enzymatic kinetics and molecular docking analysis.
Diabetes mellitus is a worldwide chronic metabolic disease that causes a major challenge for the health-care system. The high prevalence of diabetes has accelerated the search for novel therapeutic alternatives.1 At present, improving postprandial hyperglycemia is a key strategy for the treatment of diabetes and its complications.2 α-Glucosidase inhibitors (AGIs), which prevent postprandial hyperglycemia by delaying the digestion of carbohydrates in the gut, are usually employed for controlling postprandial blood glucose levels. Unfortunately, utilization of clinical AGIs (acarbose, miglitol and voglibose) often leads to severe gastrointestinal adverse effects such as abdominal discomfort and flatulence.3 As a consequence, searching for natural AGIs without side-effects from natural sources has drawn much attention in the past decade.4,5
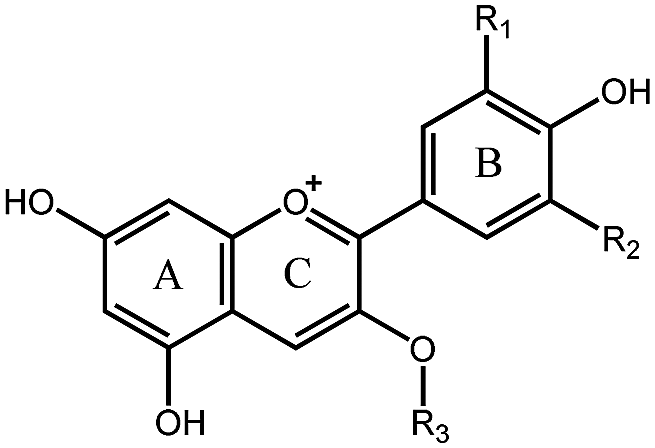
Berry fruits are regarded as a valuable food for human health.6–8 Accumulating evidence unveiled that consumption of berry extracts alleviated hyperglycemia in diabetes by inhibition of digestion enzymes and improvement of insulin resistance.9,10 Our previous work also confirmed that oral administration of mulberry anthocyanin extracts improved insulin resistance in db/db mice.11 Recently, we found that berry anthocyanin extracts possess potent α-glucosidase inhibitory activity (Fig. S1, ESI†), giving us an implication that anthocyanins may be considered as a new class of α-glucosidase inhibitors. To date, there has been no systematic report on the structure–activity relationships (SAR) for inhibition of α-glucosidase by natural anthocyanins. The possible reason may be attributed to the difficulty in obtaining monomeric anthocyanins. The flavylium cation in the anthocyanin structure (Table 1) significantly increases the difficulty of chemical synthesis. Consequently, isolation of anthocyanins from plants becomes a major approach to obtain pure monomeric anthocyanins.
|
|
|||||
|---|---|---|---|---|---|
| Number | Compound | R1 | R2 | R3 | IC50 (μM) |
| 1 | Pg3R | H | H | Rutinoside | 1.69 |
| 2 | Pg3G | H | H | Glucoside | 10.35 |
| 3 | C3R | OH | H | Rutinoside | 608.69 |
| 4 | C3G | OH | H | Glucoside | 1025.32 |
| 5 | C3S | OH | H | Sambubioside | >4000 |
| 6 | D3A | OH | OH | Arabinoside | 22.70 |
| 7 | D3R | OH | OH | Rutinoside | 69.82 |
| 8 | D3G | OH | OH | Glucoside | 142.68 |
| 9 | D3Ga | OH | OH | Galactoside | 148.20 |
| 10 | D3S | OH | OH | Sambubioside | >4000 |
| 11 | Pet3A | OCH3 | OH | Arabinoside | 81.47 |
| 12 | Pet3G | OCH3 | OH | Glucoside | 158.65 |
| 13 | Pet3Ga | OCH3 | OH | Galactoside | 234.40 |
| 14 | M3A | OCH3 | OCH3 | Arabinoside | 11.32 |
| 15 | M3G | OCH3 | OCH3 | Glucoside | 116.77 |
| 16 | M3Ga | OCH3 | OCH3 | Galactoside | 139.64 |
| 17 | Peo3A | OCH3 | H | Arabinoside | 18.75 |
| 18 | Peo3Ga | OCH3 | H | Galactoside | 143.31 |
| 19 | Acarbose | — | — | — | 356.26 |
Herein, we developed an effective protocol to obtain monomeric anthocyanin from natural sources, and then investigated the α-glucosidase inhibitory activity of the natural anthocyanins, as well as the SAR. To the best of our knowledge, this is the first study to systematically report the SAR for inhibition of α-glucosidase by natural anthocyanins.
In order to clearly compare the inhibitory activity of monomeric anthocyanin against α-glucosidase, anthocyanin purification was performed. The separation of monomeric anthocyanin analogs from natural sources via a single chromatographic technique is quite difficult, due to their closely related structures. Interestingly, some novel retention characteristics of anthocyanins in a HSCCC system were firstly found. For HSCCC purification, separation efficiency depends on the fast partitioning effect of analytes in the two-phase solvent system. Generally, a compound with a smaller partition coefficient (K) elutes faster.12 The K values of berry anthocyanins in the two-phase solvent system of n-butanol–tert-butyl methyl ether–acetonitrile–water–trifluoroacetic acid (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1:5:0.01, v/v) were measured (Table S2, ESI†). The elution order of anthocyanins was found to be more closely related to glycoside than aglycone. Anthocyanins linked with disaccharides were eluted earlier than monosaccharides in the HSCCC system. For example, D3S and C3S were eluted first. Anthocyanins linked with the same glycoside were eluted together except pelargonidin anthocyanins, such as D3Ga, Pet3Ga, M3Ga and Peo3Ga. According to our results, the elution order of anthocyanins was sambubioside, rutinoside, galactoside, glucoside and arabinoside. As for anthocyanins with the same glycoside, they followed the order of delphinidin, petunidin, malvidin, cyanidin, peonidin and pelargonidin. The elution order of anthocyanins by HSCCC turned out to be totally different from that by HPLC. For conventional HPLC purification, a high-polarity compound elutes faster than a low-polarity one, as a result, the elution of anthocyanins followed the order of delphinidin, cyanidin, petunidin, peonidin, pelargonidin and malvidin. On the basis of these novel characteristics, we developed an effective protocol (Fig. S3, ESI†) to isolate monomeric anthocyanin by a combination of HPLC and HSCCC techniques. The detailed description about the protocol is shown in Section 1 of the ESI.† 15 monomeric anthocyanins with a purity of at least 95% (HPLC) were isolated from the six kinds of berry fruit (strawberry, raspberry, blueberry, mulberry, bayberry and cranberry) through this protocol. In addition, other types of food materials, including roselle and eggplant, were employed to test the universality of this protocol (Fig. S4, ESI†). Three major anthocyanins (D3S, C3S and D3R) with a purity over 95% were obtained successfully. The structures and purities of the obtained anthocyanins were elucidated by NMR and HPLC (Fig. S6–S23, ESI†). After these tests, the proposed protocol was expected to be a universal and promising approach for the purification of anthocyanins from natural sources.
:2:1:5:0.01, v/v) were measured (Table S2, ESI†). The elution order of anthocyanins was found to be more closely related to glycoside than aglycone. Anthocyanins linked with disaccharides were eluted earlier than monosaccharides in the HSCCC system. For example, D3S and C3S were eluted first. Anthocyanins linked with the same glycoside were eluted together except pelargonidin anthocyanins, such as D3Ga, Pet3Ga, M3Ga and Peo3Ga. According to our results, the elution order of anthocyanins was sambubioside, rutinoside, galactoside, glucoside and arabinoside. As for anthocyanins with the same glycoside, they followed the order of delphinidin, petunidin, malvidin, cyanidin, peonidin and pelargonidin. The elution order of anthocyanins by HSCCC turned out to be totally different from that by HPLC. For conventional HPLC purification, a high-polarity compound elutes faster than a low-polarity one, as a result, the elution of anthocyanins followed the order of delphinidin, cyanidin, petunidin, peonidin, pelargonidin and malvidin. On the basis of these novel characteristics, we developed an effective protocol (Fig. S3, ESI†) to isolate monomeric anthocyanin by a combination of HPLC and HSCCC techniques. The detailed description about the protocol is shown in Section 1 of the ESI.† 15 monomeric anthocyanins with a purity of at least 95% (HPLC) were isolated from the six kinds of berry fruit (strawberry, raspberry, blueberry, mulberry, bayberry and cranberry) through this protocol. In addition, other types of food materials, including roselle and eggplant, were employed to test the universality of this protocol (Fig. S4, ESI†). Three major anthocyanins (D3S, C3S and D3R) with a purity over 95% were obtained successfully. The structures and purities of the obtained anthocyanins were elucidated by NMR and HPLC (Fig. S6–S23, ESI†). After these tests, the proposed protocol was expected to be a universal and promising approach for the purification of anthocyanins from natural sources.
After purification, 18 monomeric anthocyanins involving six kinds of anthocyanidin and five forms of glycosyl moiety were used for investigating the SAR of anthocyanins against α-glucosidase. The activities of anthocyanins followed certain patterns (Table 1), thus ranking of the activities of anthocyanidin aglycones as well as glycosyl groups (glycosides) was proposed. As for aglycones, the facts that Pg3G (IC50 = 10.35 μM) > M3G (IC50 = 116.77 μM) > D3G (IC50 = 142.68 μM) > Pet3G (IC50 = 158.65 μM) > C3G (IC50 = 1025.32 μM) and M3A (IC50 = 11.32) > Peo3A (IC50 = 18.75) > D3A (IC50 = 22.70) > Pet3A (IC50 = 81.47) led us to propose that pelargonidin (Pg) > malvidin (M) > peonidin (Peo) > delphinidin (D) > petunidin (Pet) > cyanidin (C). The results that Pg3R (1.69 μM) > D3R (69.82 μM) > C3R (608.69 μM) and M3Ga (139.64 μM) > Peo3Ga (143.31 μM) > D3Ga (148.20 μM) > Pet3Ga (234.40 μM) further confirmed the ranking of the activities of anthocyanidin aglycones (Pg > M > Peo > D > Pet > C). As for glycosyl moieties, based on the examined activities of Pg3R > Pg3G, C3R > C3G and D3R > D3G, we assumed that rutinoside > glucoside. Considering the inhibitory activities of M3A > M3G > M3Ga, Pet3A > Pet3G > Pet3Ga and D3A > D3G > D3Ga, we proposed the ranking of these three sugar groups to be arabinoside > glucoside > galactoside. In combination of the results that D3A > D3R and C3S (IC50 > 4000 μM) and D3S (IC50 > 4000 μM) both showed the lowest inhibitory effects, we assumed the ranking of the glycosyl groups to be arabinoside > rutinoside > glucoside > galactoside > sambubioside.
Among these anthocyanins, Pg3R showed the most potent α-glucosidase inhibitory activity. Therefore, Pg3R was selected as a representative anthocyanin for investigating its inhibition mechanism. Lineweaver–Burk analysis suggested that Pg3R induced a mixed type of inhibition against α-glucosidase (Fig. 1a). The interaction between Pg3R and α-glucosidase was subsequently investigated by fluorescence spectroscopy and circular dichroism (CD) spectroscopy. As shown in Fig. 1b, the intrinsic fluorescence intensity of α-glucosidase decreased gradually with the increased concentrations of Pg3R, indicating that Pg3R interacted with α-glucosidase and then quenched its intrinsic fluorescence. The quenching rate constants (Kq) at the incubation temperatures of 291 K, 303 K, and 310 K were much larger than the maximum scattering collision quenching constant of the biomacromolecule (2 × 1010 L mol−1 s−1), demonstrating that static quenching dominated the fluorescence quenching process (Table S3, ESI†). The number of binding sites (n) at the three incubation temperatures was all close to one, indicating that Pg3R has only one binding site to interact with α-glucosidase. The binding constants (Ka) at the three temperatures were in the order of 104 L mol−1, indicating that a high binding affinity existed in the complex of Pg3R with α-glucosidase. In addition, the thermodynamic parameters (ΔS, ΔH and ΔG) were calculated. We found that ΔH and ΔS were positive, while ΔG was negative (Table S3, ESI†). According to the theory of Ross and Subramanian (namely, relative values’ change in ΔH and ΔS),13 it can be concluded that the binding process was spontaneous, which was driven mainly by a hydrophobic force. CD spectroscopy analysis showed that the absorption of the two negative peaks at 209 and 222 nm decreased after the addition of 50 μM or 100 μM of Pg3R, which demonstrated a loss of the α-helix structure (Fig. 1c). With an increase in molar ratios of Pg3R to α-glucosidase (from 36:1 to 72:1), the percentages of the α-helix structure and β-turn decreased from 22.7% to 17.1% and from 11.9% to 7.3%, respectively, whereas the content of the β-sheet increased from 34.2% to 47.1% (Table S4, ESI†). The transformation from the α-helix to the β-sheet conformation in the presence of Pg3R indicated a partial unfolding of the α-glucosidase structure, which might destroy some hydrogen bonding networks and cause alterations of the secondary structure of α-glucosidase. These alterations may prevent the binding of the substrate to α-glucosidase or hamper the formation of an active center, eventually leading to the inactivation of the enzyme. In order to confirm that the alteration of α-helix is associated with the inhibitory activity of α-glucosidase, the effects of other representative anthocyanins (Pg3G, M3A and D3S) on the secondary structure of α-glucosidase were investigated (Fig. 1d–f). The results showed that two anthocyanins (Pg3G and M3A) with potent inhibitory activity (IC50 < 15 μM) remarkably decreased the percentage of α-helix of α-glucosidase, while D3S with poor inhibitory activity (IC50 > 4000 μM) slightly decreased the content of α-helix (Table S4, ESI†). These results jointly confirmed that the decreased α-glucosidase activity was associated with the alteration of the α-helix structure.
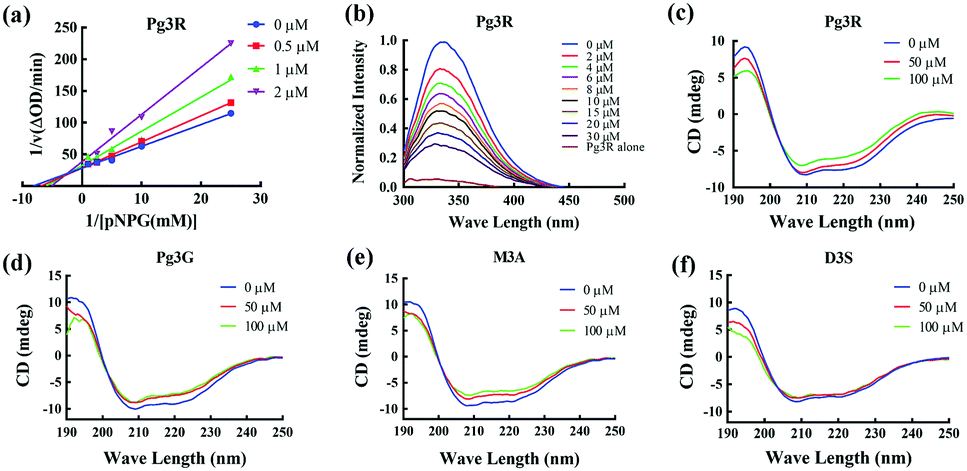 | ||
| Fig. 1 Inhibition mechanism of Pg3R and its analogs against α-glucosidase. Lineweaver–Burk plot of Pg3R (a). Fluorescence spectrum of Pg3R with α-glucosidase (b). CD spectra of Pg3R with α-glucosidase (c), Pg3G with α-glucosidase (d), M3A with α-glucosidase (e) and D3S with α-glucosidase (f). | ||
To provide deep insight into the interaction of Pg3R with α-glucosidase (MAL12), a molecular docking study was conducted. We constructed a homology model whose quality was verified by passing both Ramachandran plot assessment (with 97.3% of residues in the favored region and 2.7% of residues in the allowed region) and 3D tests (with a score of 92.29%). This model was then used for docking with Pg3R and its analogs. The proposed binding pose of Pg3R indicated that it could fit well into the binding pocket of the homology model, with a predicted binding energy of −16.6 kcal mol−1 (Fig. S5, ESI†). Asp214, Glu276 and Asp349 were reported to be the key residues that are responsible for the catalytic activities of α-glucosidase, and interactions formed with them were proposed to be important features of the inhibitory capacities of small molecular ligands.14,15 As we can see from Fig. 2, Pg3R was supposed to have electrostatic (π–anion) interactions with the residues Asp214, Glu276, and Asp349, along with forming one hydrogen bond with Glu276. Pg3R was also predicted to form hydrogen bonds with Asp68, Ser156, His279, Arg312, Asp408, Asn412, and Arg439, as well as to have hydrophobic interactions with Tyr71 (π–π), Ala278 (π–alkyl) and His279 (π–alkyl). To further illustrate the important interactions that are responsible for the catalytic activities of α-glucosidase, four anthocyanins with potent inhibitory activity (M3A, Peo3A, D3A and Pg3G, IC50 < 30 μM) and four anthocyanins with poor activity (C3S, D3S, C3G and C3R, IC50 > 600 μM) were docked with α-glucosidase. The results showed that the A ring and C ring of M3A, Peo3A, D3A and Pg3G could all enter in depth of the active pocket (Fig. 3) and form hydrophobic interactions with Glu276, Asp214, Ala278, Tyr71, Asp349 residues (Fig. 2). Besides, their glycosyl groups were all interacted with Asp408 via hydrogen bonds. However, for those anthocyanins with poor activity, their aglycones could not enter in depth of the active pocket, especially for D3S and C3S (Fig. 3), and as a consequence, the abovementioned interactions disappeared. On the basis of these results, it can be concluded that the hydrophobic interactions formed between aglycones and active pocket residues (Glu276, Asp214, Ala278, Tyr71, and Asp349) as well as the hydrogen bond formed between glycoside and Asp408 and Glu276 could enhance the α-glucosidase inhibitory activity of anthocyanins.
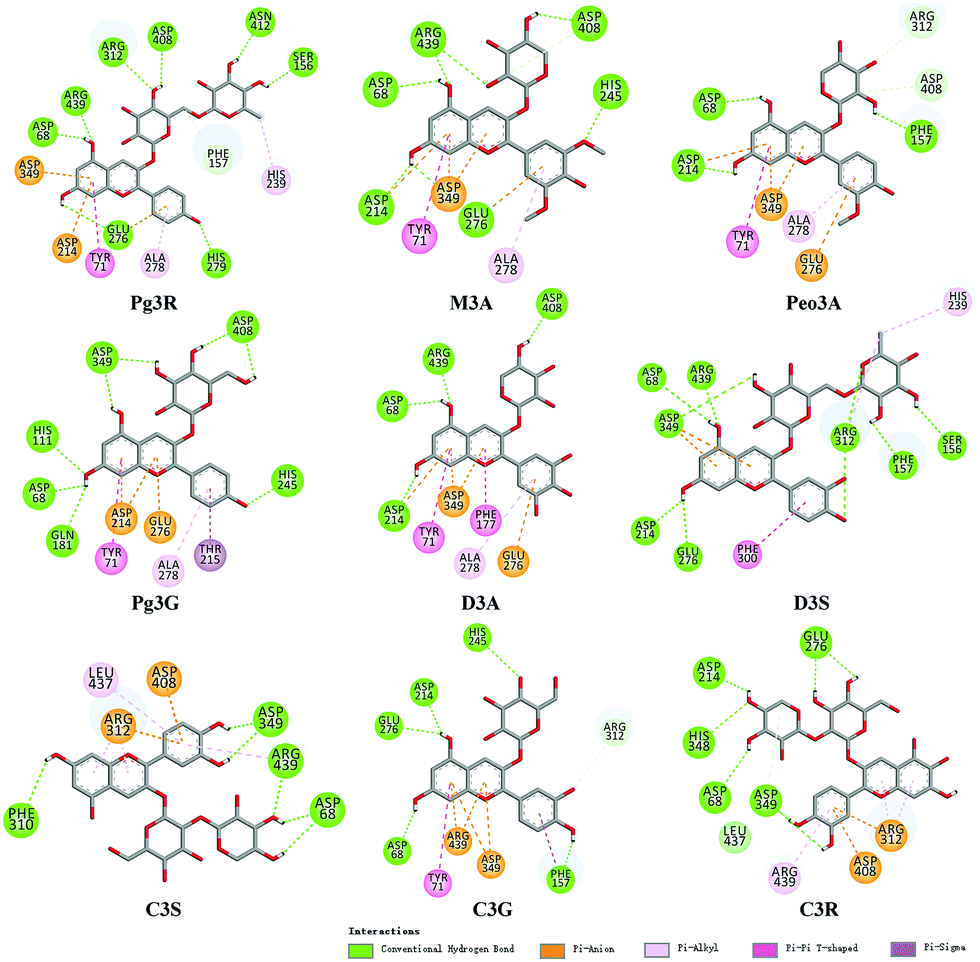 | ||
| Fig. 2 Molecular docking of Pg3R and its analogs with α-glucosidase; interaction of anthocyanins with α-glucosidase. | ||
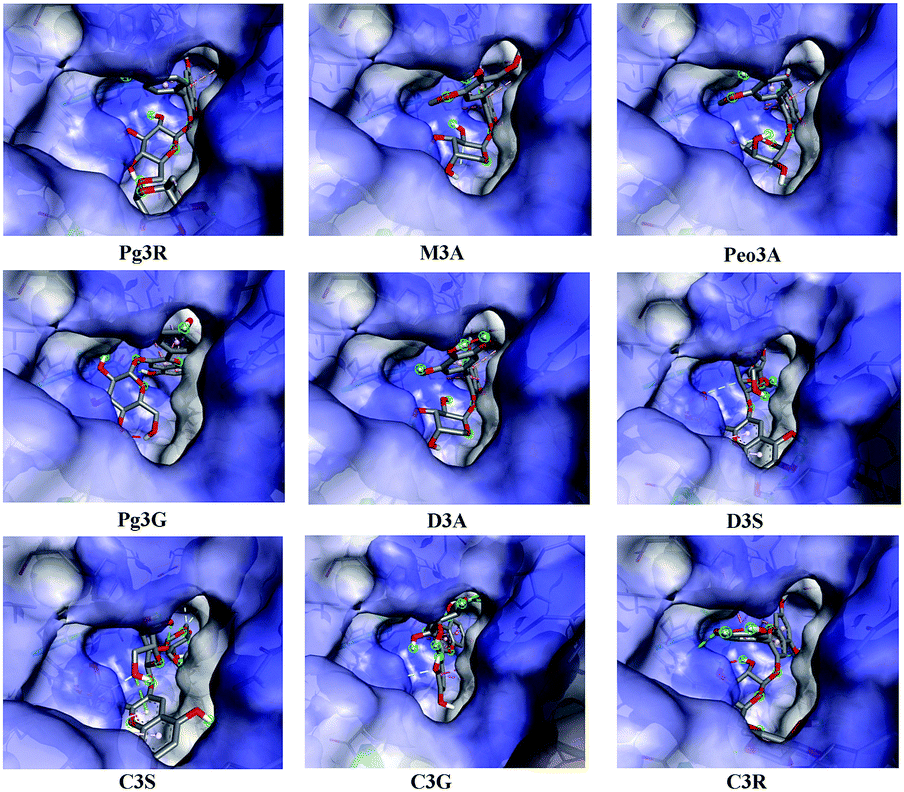 | ||
| Fig. 3 Proposed binding pose of anthocyanins to the active pocket of α-glucosidase. | ||
To further investigate the intestinal α-glucosidase inhibitory activity in vivo, oral sucrose and oral maltose tolerance tests in a normal ICR mice model were performed. Acarbose at doses of 25 mg kg−1 BW and 50 mg kg−1 BW was used as a positive control. In the oral sucrose tolerance test (Fig. 4a), a rapid increase in the blood glucose level from 5.28 ± 0.59 mM to a maximum of 11.2 ± 0.75 mM in 30 min after oral administration of sucrose (2 g kg−1 of BW) was observed for the negative control group. After that the blood glucose concentration recovered to the pretreatment level at 120 min. As for the drug-treatment group, Pg3R significantly reduced the blood glucose levels at 15 min and 30 min when compared with that of the negative control group, and the suppression of the blood glucose levels was in a dose-dependent manner. Moreover, treatment with Pg3R led to 18.5% decrease of the AUC at a dose of 50 mg kg−1 BW and 32.5% decrease at a dose of 150 mg kg−1 BW, with the effects observed comparable to those of acarbose at doses of 25 mg kg−1 BW (23.3% decrease) and 50 mg kg−1 BW (24.7%) (Fig. 4b). Subsequently, a maltose tolerance test was carried out for comparison. Similar to the sucrose tolerance test, Pg3R treatment provoked a significant decrease of the postprandial peak in a dose-dependent manner compared with that of the negative control group. The AUC for postprandial plasma glucose in 2 h after Pg3R administration was reduced by 15.3% at a dose of 50 mg kg−1 BW and 28.4% at a dose of 150 mg kg−1 BW, compared with that of the negative control (Fig. 4c and d). The good activity observed during the oral sucrose and maltose tolerance tests strongly confirmed that Pg3R could suppress the postprandial hyperglycemia by inhibiting intestinal α-glucosidase. Therefore, natural Pg3R can be regarded as a promising candidate for α-glucosidase inhibitors.
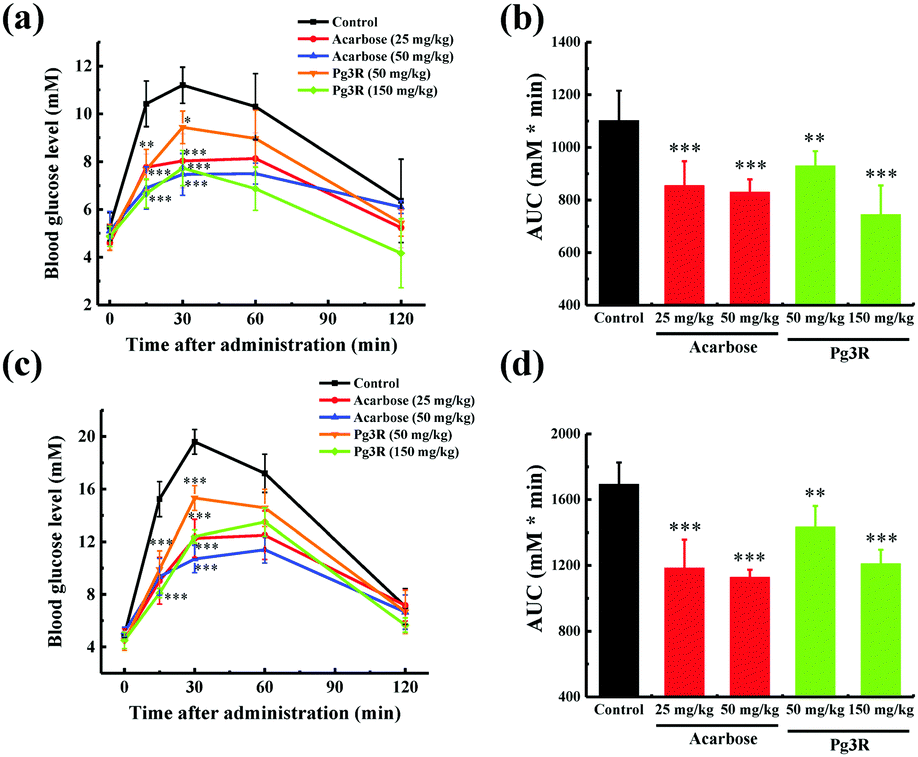 | ||
| Fig. 4 Effects of acarbose and Pg3R on postprandial blood glucose levels in normal ICR mice. (a) Blood glucose concentrations after oral administration of Pg3R and sucrose. (b) Area under the curve (AUC) after oral administration of Pg3R and sucrose for 2 h. (c) Blood glucose concentrations after an oral administration of Pg3R and maltose. (d) Area under the curve (AUC) after oral administration of Pg3R and maltose for 2 h. Each value represents the mean ± standard deviation (n = 9). Asterisks indicate a significant difference (*p < 0.05, **p < 0.01, and ***p < 0.001) compared with the control group. | ||
In summary, our study identified a potent α-glucosidase inhibitor (Pg3R) that can improve postprandial hyperglycemia, and for the first time provides a deep insight into the SAR of α-glucosidase inhibitory activity of anthocyanins. Also, we developed an effective protocol to isolate monomeric anthocyanins from natural sources, which is beneficial for expanding the market of anthocyanin standards as well as promoting research on its biological activities.
This work was supported by grants from the Zhejiang Provincial Natural Science Foundation of China (LR18C200002) and the National Natural Science Foundation of China (21876152 and U1703105).
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. K. Ghosh, B. S. Reddy, Y. C. Yen, E. Cardenas, K. V. Rao, D. Downs, X. Huang, J. Tang and A. D. Mesecar, Chem. Sci., 2016, 7, 3117–3122 RSC.
- A. Ceriello, Diabetes, 2005, 54, 1–7 CrossRef CAS.
- N. A. Calcutt, M. E. Cooper, T. S. Kern and A. M. Schmidt, Nat. Rev. Drug Discovery, 2009, 8, 417–429 CrossRef CAS.
- Y.-T. Deng, S.-Y. Lin-Shiau, L.-F. Shyur and J.-K. Lin, Food Funct., 2015, 6, 1539–1546 RSC.
- S. Deng, L. Xia and H. Xiao, Chem. Commun., 2014, 50, 2582–2584 RSC.
- W. Chen, H. Su, Y. Xu, T. Bao and X. Zheng, Food Chem., 2016, 196, 943–952 CrossRef CAS PubMed.
- Y. Li, T. Bao and W. Chen, Food Chem., 2018, 243, 65–73 CrossRef CAS PubMed.
- L. Xie, H. Su, C. Sun, X. Zheng and W. Chen, Trends Food Sci. Technol., 2018, 72, 13–24 CrossRef CAS.
- A. S. Boath, D. Stewart and G. J. McDougall, Food Chem., 2012, 135, 929–936 CrossRef CAS.
- V. Gowd, Z. Jia and W. Chen, Trends Food Sci. Technol., 2017, 68, 1–13 CrossRef CAS.
- F. Yan, G. Dai and X. Zheng, J. Nutr. Biochem., 2016, 36, 68–80 CrossRef CAS.
- Y. Ito, J. Chromatogr. A, 2005, 1065, 145–168 CrossRef CAS.
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20, 3096–3102 CrossRef CAS.
- G. Wang, D. He, X. Li, J. Li and Z. Peng, Bioorg. Chem., 2016, 65, 167–174 CrossRef CAS.
- K. Wang, L. Bao, K. Ma, J. Zhang, B. Chen, J. Han, J. Ren, H. Luo and H. Liu, Eur. J. Med. Chem., 2017, 127, 1035–1046 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc07985d |
| This journal is © The Royal Society of Chemistry 2019 |