
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEn route to CO2-containing renewable materials: catalytic synthesis of polycarbonates and non-isocyanate polyhydroxyurethanes derived from cyclic carbonates
Felix D.
Bobbink
 *,
Antoine P.
van Muyden
and
Paul J.
Dyson
*,
Antoine P.
van Muyden
and
Paul J.
Dyson
Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland. E-mail: felix.bobbink@epfl.ch
First published on 18th December 2018
Abstract
Combining CO2-chemistry with biomass conversion allows renewable polymeric materials including polycarbonates and polyhydroxyurethanes (PHUs) to be generated. The demand for robust materials with modular properties that can be prepared on an industrial scale is important and, to date, the most important polymeric materials are derived from petrochemicals. These materials inevitably result in CO2 emissions, and therefore making robust materials from renewable sources will contribute to a more sustainable society. An attractive way to address this challenge is to combine biomass transformations with CO2-fixation and material science. An identified target that combines all three aspects involves the preparation of PHUs (or non-isocyanate polyurethanes, NIPUs) via the polymerization of fully renewable cyclic carbonates derived from biomass and CO2 with a diamine compound that can also been derived from biomass sources. In this review, we critically analyze the progress in catalyst development for the efficient transformation of epoxides and CO2 to cyclic carbonates and polycarbonates. We also discuss the synthesis of PHUs from cyclic carbonates and diamines (not restricted to fully renewable compounds), including challenges in regiocontrol and biodegradability, as well as the role catalysts play in the synthesis of these polymers.
 Felix D. Bobbink | Felix Bobbink obtained his PhD from the École Polytechnique Fédérale de Lausanne (EPFL) in 2018, and is currently a post doc in Prof. Paul Dyson's laboratory at the EPFL. During his PhD, he developed ionic polymers and evaluated them as catalysts for CO2-related transformations. His current work involves the preparation of polymers from cyclic carbonates and amines. |
 Antoine P. van Muyden | Antoine van Muyden obtained his MSc from the École Polytechnique Fédérale de Lausanne (EPFL) in 2016 and is currently a PhD student under the supervision of Prof. Paul Dyson at the EPFL. His work involves the preparation of heterogeneous bimetallic catalysts and their applications for biomass and CO2 tranformations. |
 Paul J. Dyson | Paul Dyson joined the Swiss Federal Institute of Technology in Lausanne (Ecole Polytechnique Fédérale de Lausanne, EPFL) in 2002. He grew up in the UK, completing his doctoral thesis at the University of Edinburgh. Prior to his appointment at the EPFL he held appointments at Imperial College of Science, Technology and Medicine and the University of York as a Royal Society University Research Fellow. |
Introduction
Today, most of society's chemicals, including chemical building blocks, fine chemicals, fuels and materials are derived from the petrochemical industry.1 The intensive utilization of fossil resources has strongly impacted on the modernization of our society and our quality of life. However, the exploitation of these resources has also led to an imbalance in the carbon cycle and other environmental problems.2,3 CO2 is naturally involved in the carbon cycle, which balances the CO2 in natural processes.4 The concentration of CO2 in the atmosphere before the industrial revolution was around 280 ppm and reached 410 ppm in July 2018. Human emissions of CO2 have stabilized over the last few years at ca. 34 GTons. CO2 acts as a greenhouse gas, which results in the warming of the earth, with potential catastrophic consequences.5 Consequently, governments have recommended that the rate of emissions should be limited in order to contain global warming to 2 °C (decided in November 2015 at COP21 in Paris).6 To achieve this goal, emissions should be limited in all possible areas of activity including electricity generation, heating, transport, construction and chemical production. This prerequisite rationalizes the efforts that are made in clean energy storage and production,7 direct CO2 reduction to fuels,8etc. Moreover, post-combustion technologies that aim at sequestering CO2 at the source of emission have emerged.9–11 These technologies are being implemented at the exhaust of power stations and rely, for example, on amine compounds that effectively form carbamate salts upon exposure to CO2. Simultaneously, other technologies are emerging that aim to capture CO2 directly from air, for example by relying on the affinity of atmospheric CO2 with bases such as metal hydroxides or amines.12,13 The aforementioned technologies are meant to capture CO2 and then release it in pure form so that it can be stored underground or under the oceans.14It would be advantageous to benefit from the CO2 that is emitted to generate value-added products, as is the case for the synthesis of urea, which is prepared from CO2 on an industrial scale.15,16 A remarkable example of CO2 valorization as a chemical reagent (or C1 source) involves the cycloaddition of CO2 into epoxides (CCE reaction) to afford cyclic organic carbonates (COCs). This reaction was discovered in the early 1950s, has been industrialized,17,18 and requires a catalyst (see below).19 The production of the smallest COCs, propylene carbonate (PC) and ethylene carbonate (EC), is particularly important because of their diverse applications and low toxicity.17,18,20 They find applications in cosmetics, as solvents,21 and in batteries,22 and, importantly, they are biodegradable.23 The reaction between epoxides and CO2 is not restricted to the formation of the smallest COCs, and various terminal epoxides have been employed.24,25 Some of these epoxides can be derived from renewable sources such as cellulose, lignin, or vegetable oils and can subsequently be processed to polycarbonates (pCs), polyurethanes (PUs) and polyhydroxyurethanes (PHUs). In PUs the monomers are linked one to another via a urethane functional group. Typically, these are formed by reacting an isocyanate with a diol (or alternatively a diamine or an aminol) and have a rich history since their development in the 1930s.26 The market for these compounds is enormous,27,28 and currently all the precursors are derived from petrochemical sources. Moreover, the isocyanate group is moisture sensitive,29 and isocyanates are notoriously toxic with, for example, the release of methyl isocyanate being responsible for the Bhopal catastrophe in India.30 Therefore, finding alternatives to existing routes is ongoing, and routes involving bio-based or partially bio-based PUs/PHUs are under investigations.31–35 Efforts to discover novel materials that are derived from biomass sources and that display properties comparable to those of existing materials is important en route towards a more sustainable chemical industry and society.
In this review, we will present the state of catalyst research for the preparation of cyclic organic carbonates (COCs) and their corresponding polycarbonates. In addition, the synthesis of PHUs from COCs and diamines will be described and compared to the well-established PU synthesis. Throughout this review, the focus is directed to the available catalytic tools, the renewable aspects of the reaction and the biodegradability of the materials.
Role of catalysis in CO2-containing COCs and pCs
COC synthesis
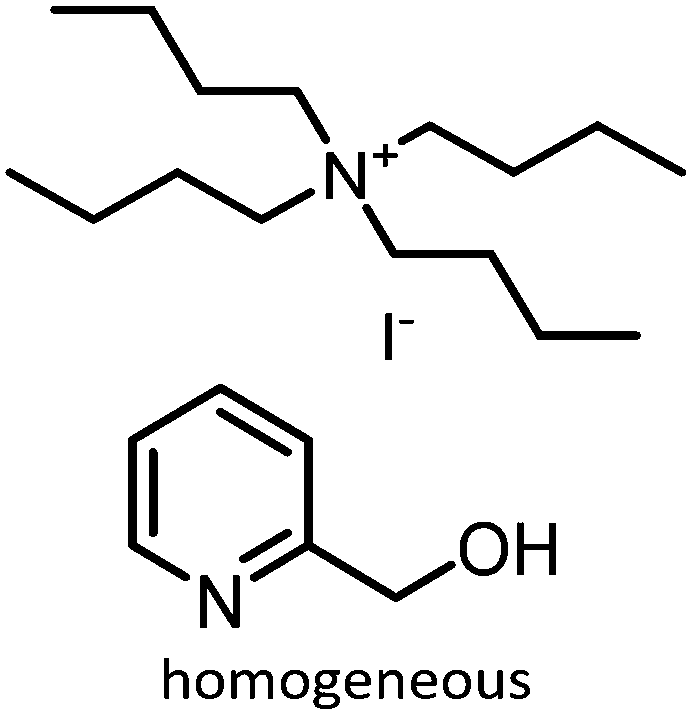
Prior to discussing the synthesis of COCs from bio-based epoxides some general remarks on the catalyzed CO2-epoxide coupling (CCE) reaction are provided. The CCE reaction proceeds according to Fig. 1 and represents a successful application of CO2 chemistry.36 As mentioned above, the reaction has been intensively investigated, and numerous efficient catalysts have been developed and described.37–40 For example, ionic liquids (ILs) have been identified as potent catalysts for this reaction that operate under mild, solvent-free conditions. Typical examples involve simple ammonium salts such as N-tetrabutylammonium bromide/iodide (Fig. 1, catalyst 1) or 1-butyl-3-methylimidazolium chloride (Fig. 1, catalyst 3). Similarly, alkali metal salts combined with a polar aprotic solvent have also been used (Fig. 1, catalyst 5).19,41–43 For processing purposes, heterogeneous versions of the catalysts have been developed, such as imidazolium-based polymers used in place of ILs (see Fig. 1, catalysts 2 and 6).44–47 It should be noted that while the reaction is considered “green”, little emphasis has been put on the sustainability of the catalyst itself. In this context, it has been shown that ILs are not necessarily biodegradable and that their synthesis poses problems.48–50 ILs often require dichloromethane, tetrahydrofuran (THF) or diethyl ether during synthesis, all of which are towards the bottom of the solvent sustainability scale.51 Note, ILs based on naturally occurring cations can potentially be obtained from renewable sources.52 In contrast to organocatalysts, metal composites are not derived from petrochemical resources even if their extraction can produce a significant amount of waste. These catalysts are based on available materials and can, to some extent, be considered green (Fig. 1, catalyst 4).52,53 Advances in the catalysts and the mechanism of the reaction have been reviewed several times in recent years.54–56 The CCE reaction is often considered a reaction that requires harsh conditions to proceed, but recent studies have shown that simple and cheap catalysts convert epoxides into cyclic carbonates under atmospheric pressure of CO2 at temperatures well below 100 °C.57 Notably, a study from 1993 demonstrated that the reaction takes place at atmospheric pressure.58 The mechanism of the transformation proposed for IL and organic salt catalysts proceeds via ring opening of the epoxide (Fig. 1, step 1) by the anion of the salt, followed by insertion of CO2 into the alkoxide (Fig. 1, step 2). Finally, the product is formed via a ring-closing step (Fig. 1, step 3).59 The mechanism and the rate determining step can vary according to the nature of the catalyst.60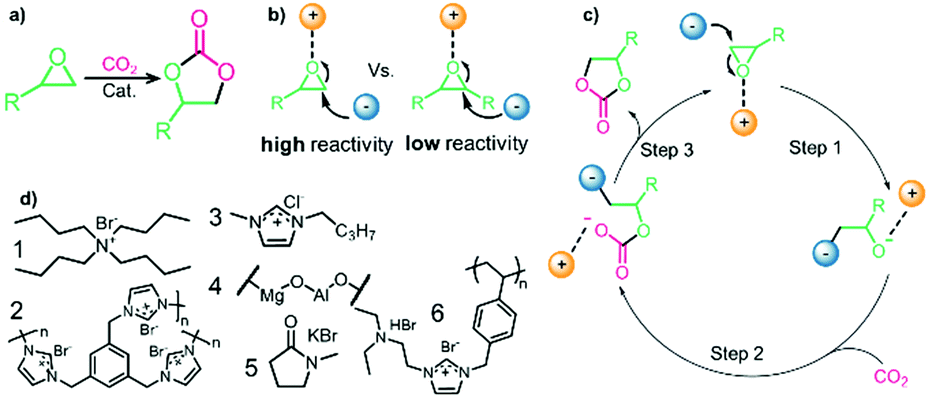 | ||
| Fig. 1 (a) General CCE reaction. (b) Key mechanistic step for IL catalyzed CCE reaction. (c) Mechanism for IL-catalyzed reaction. (d) Selected catalysts. | ||
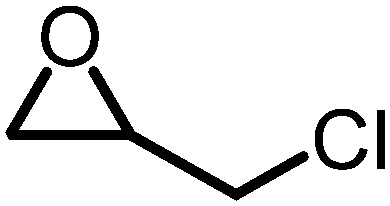
The reactivity of epoxides with CO2 is dependent upon the structure of the epoxide with the typical reactivity of different epoxides depicted in Fig. 2. Epichlorohydrin (ECH) is among the most reactive epoxides for the CCE reaction (converted to (chloromethyl)ethylene carbonate, which will be referred to as epichlorohydrin carbonate, EpC) and can be converted at 50 °C under 1 atm of CO2 using a simple homogeneous IL catalyst.57 It should be noted that ECH can be prepared from glycerol, a renewable chemical obtained during biodiesel production (via the Epicerol® process).61 Despite the renewable aspect of ECH, it is very toxic and must be handled with care, and this may hinder industrial development.62
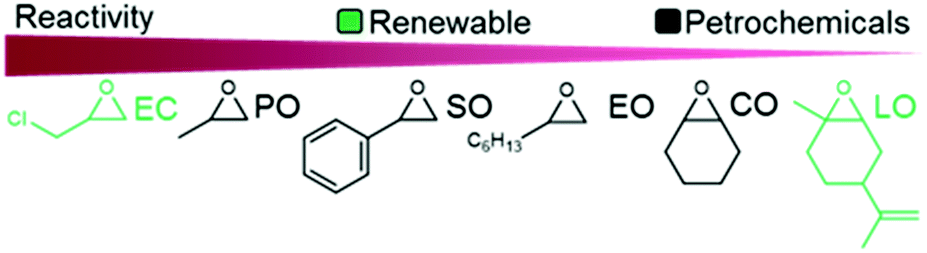 | ||
| Fig. 2 Epoxides derived from non-renewable and renewable sources and their relative reactivity for COC synthesis. | ||
Complete solubility/miscibility of the salt catalyst in the reaction solution is crucial for high activity, a feature that is frequently overlooked in organic-salt catalyzed reactions (ILs are not necessarily soluble in the epoxide under solvent-free conditions, and can therefore be considered as heterogeneous catalysts rather than homogeneous). Furthermore, propylene oxide (PO) and ethylene oxide (EO) are somewhat inconvenient to use on a laboratory scale due to their low boiling points, thus necessitating the use of pressure reactors and, consequently, they are not exclusively used as model substrates, despite their high industrial importance. The utilization of internal epoxides such as cyclohexene oxide (CyO) or limonene oxide (LO) requires harsher reaction conditions because of the lower reactivity of the tertiary carbon compared to secondary carbons in terminal epoxides (steric hindrance). Notably, both internal and terminal epoxides can be derived from renewable sources.63
Table 1 compiles reaction conditions for selected CO2-epoxide cycloadditions, including examples of homogeneous and heterogeneous salt catalyzed reactions (Table 1, entries 1, 2, 4 and 5), as well as a Mg-Al-oxide catalyst (Table 1, entry 3). The table compiles one simple organic salt (Table 1, entry 1), one heterogeneous salt (entry 2), and three examples of H-bond donor salts (entries 4–6). The example of inorganic catalyst (entry 3) is included as a reminder that many classes of catalysts have been developed over the past decades. Direct comparison between catalysts is hampered by the absence of benchmark experimental reaction conditions. Typical reaction conditions for propylene oxide conversion require pressures of 5–10 atm at temperatures ranging from room temperature to 100 °C (Table 1, entry 3). When other epoxides are employed, the reaction can conveniently be conducted at atmospheric pressure at temperatures ranging from 25 °C to 100 °C (Table 1, entries 1, 2, 4 and 5). The difficulty to compare different catalysts has been identified previously and benchmark conditions have been proposed,57,64 as it is difficult to assess the extent of progress that has been made between the catalysts reported between 1999 (Table 1, entry 3) and 2018 (Table 1, entries 4 and 5). Nonetheless, it appears that the most promising systems in terms of catalytic efficiency are composed of an onium salt combined with an external H-bond donor, allowing the reaction to proceed at room temperature under atmospheric pressure (Table 1, entry 6).57,64,65 It should be noted that the catalysts for COC synthesis presented above are most often not oxygen sensitive and the reaction does not require anhydrous conditions and water can even accelerate the reaction rates of certain IL catalysts.66 Epoxides possess a stereogenic center and hence stereoselective reactions are possible using chiral catalysts that afford optically pure COCs,67–69 although optically pure COCs may also be obtained from the catalytic chiral resolution of racemic epoxides.70
| Entry | Catalyst | Epoxide | Reaction parameters | Yield [%] | Ref. year |
|---|---|---|---|---|---|
| 1 |
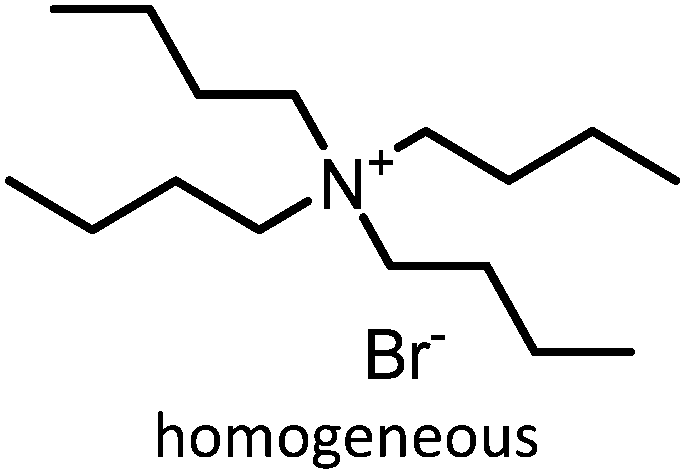
|
|
1 atm, 60 °C, 22 h, 2 mol%, 30 g epoxide | 78 | 42, 2002 |
| 2 |
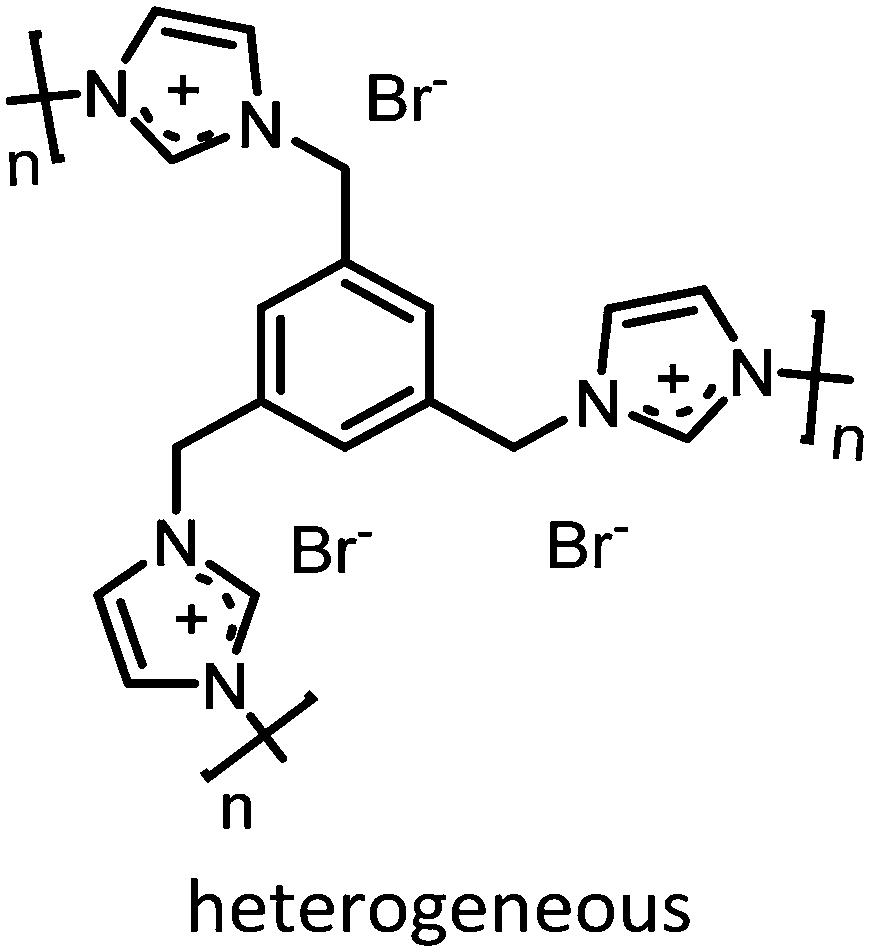
|

|
1 atm, 100 °C, 24 h, 5 mol%, 76 mg epoxide | 99 | 44, 2017 |
| 3 |
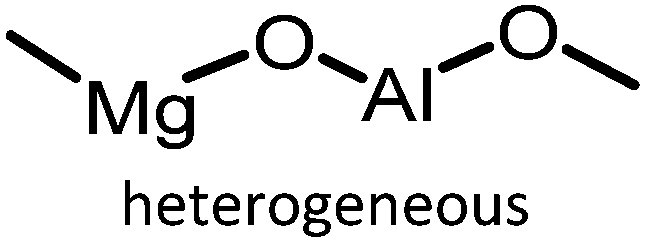
|

|
5 atm, 100 °C, 24 h, 0.5 g cat, 0.232 g epoxide | 88 | 53, 1999 |
| 4 |
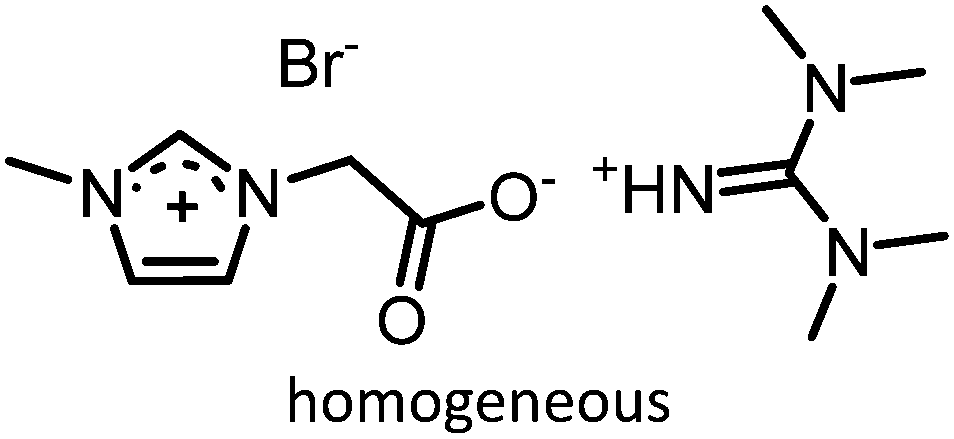
|

|
1 atm, 30 °C, 12 h, 25 mol%, 0.185 g epoxide | 84 | 71, 2018 |
| 5 |
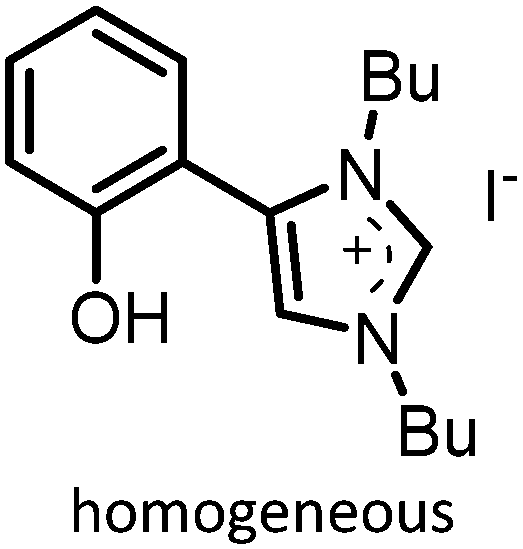
|

|
10 atm, 80 °C, 1 h, 0.75 mol% cat., 148 mg epoxide | 99 | 72, 2018 |
| 6 |
|

|
1 atm, 25 °C, 20 h, 8 mol% cat., 348 mg epoxide | 86 | 73, 2016 |
pC synthesis
Catalysts have also been developed that convert epoxides and CO2 directly into pCs. The mechanistic pathway of the reaction differs and, for the direct polymerization, a metal catalyst is usually required (see Fig. 3 for representative examples).74 The main epoxides that are converted into pCs are PO, CyO and LO and lately efforts have been devoted to the development of tailored epoxides for the reaction.75 Co-polymerization of PO with CO2 leads to polypropylene carbonate (pPC), a polymer that finds applications in ceramics (as a binder), in adhesives and as a propellant.76,77 Polymerization of CyO with CO2 leads to polycyclohexene carbonate (pCyC). The pC prepared from LO and CO2 is a viable bio-based polymer candidate for industrial applications since the resulting polymer displays excellent properties including high thermal resistance, hardness and transparency as well as having a low polydispersity index (PDI < 1.2) and high molecular weight (108.6 kDa).78 Moreover, in contrast to many other polymers (polyethylene for example), pCs are often biodegradable, a major advantage as plastic waste is heavily polluting the environment.79–82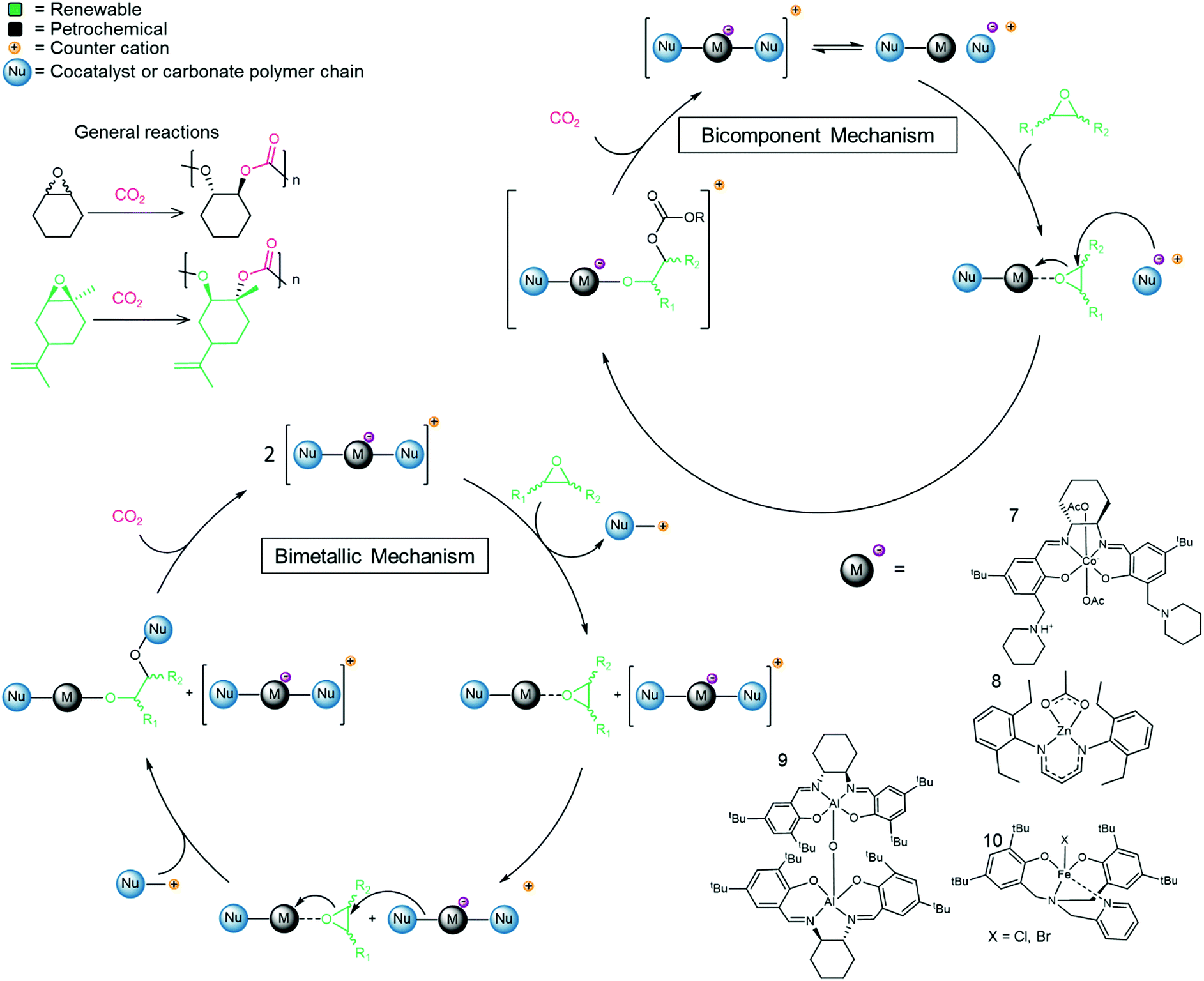 | ||
| Fig. 3 Co-polymerization of epoxides and CO2 to afford pCs, representative catalysts and the generally accepted bicomponent and bimetallic catalytic cycles.64,74,78,97,98 | ||
As discussed above, the CCE reaction to prepare COCs is most efficient with terminal epoxides, and mechanistic studies show that internal epoxides (CyO for example) result in a more controlled polymerization reaction than terminal epoxides and decreases the selectivity towards COCs.74 Furthermore, the use of CyO (or LO) facilitates co-polymerization while avoiding the formation of unwanted polyethers (pEs) derived from the epoxide.83–85 While pEs are unwanted in the context of pC synthesis, these polymers are also valuable in their own right and are obtained from the polymerization of epoxides in the absence of CO2. For instance, polypropylene oxide (pPO) is used as a component in PU synthesis.86 Efforts to produce isospecific polymers by catalytic routes are ongoing.87 Interestingly, the catalysts employed in the synthesis of pC and pE tend to be structurally related. For example, mono- and bimetallic cobalt salen complexes have been employed as effective catalysts to transform PO into pPO.88,89 Bimetallic Cr-based catalysts afford isospecific polyethers containing alcohol end-groups,90 and the resulting polymers are semi-crystalline and display average molecular weights (Mn) in the range 2–15 kDa. Active catalysts for the co-polymerization of CO2 and epoxides are typically metal-based and may contain ionic sites directly attached to the ligands.91,92 Alternatively, when no ionic part is present in the metal-catalyst, a co-catalyst is required to promote the reaction, typically TBAB or PPNCl or other salts that are commonly used in COC synthesis (see examples in Table 2). Several metal complexes have been successfully applied for this reaction including Co,74,93 Zn,94 Al64 and Cr,95,96 and examples of Fe97 and In98 complexes have also been reported (see Fig. 3, structures 7–10 as examples).99 The reaction proceeds via either a bicomponent or bimetallic pathway, and this typically depends on the catalyst and reaction parameters.100,101 In this context, evidence for a monometallic pathway has been provided for an In catalyst (see Fig. 3 for general catalytic cycle).98 Recently, an example of a metal-free, salt catalyst has been reported for the polymerization reaction, which is of interest towards the development of a sustainable, metal-free process.102
| Entry | Catalyst | Epoxide | Reaction parameters | Remarks | Ref, year |
|---|---|---|---|---|---|
| 1 |
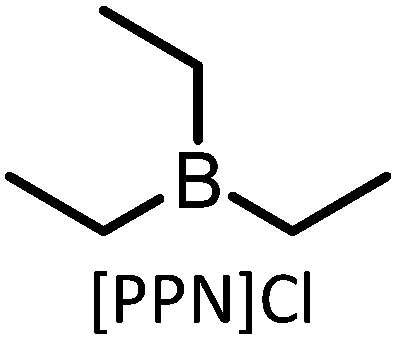
|
|
10 atm, 60 °C, 10 h, THF solvent | Yield = 35%, Mn = 37 kDa, PDI = 1.1 | 102, 2016 |
| 2 |
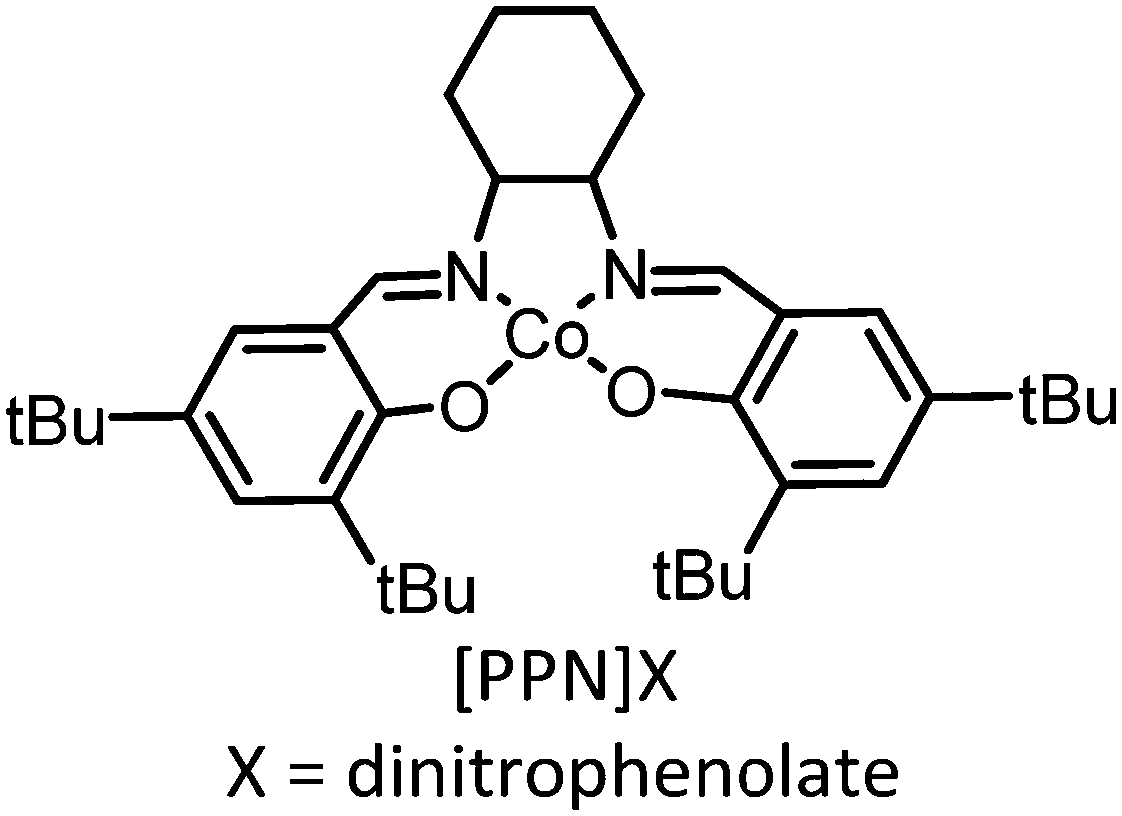
|
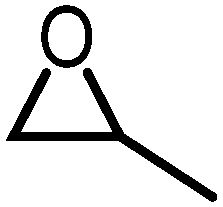
|
15 atm, 22 °C, 2 h | TON = 802 ± 26, Mn = 27 kDa, oxidant = FcPF6 | 113, 2016 |
| 3 |
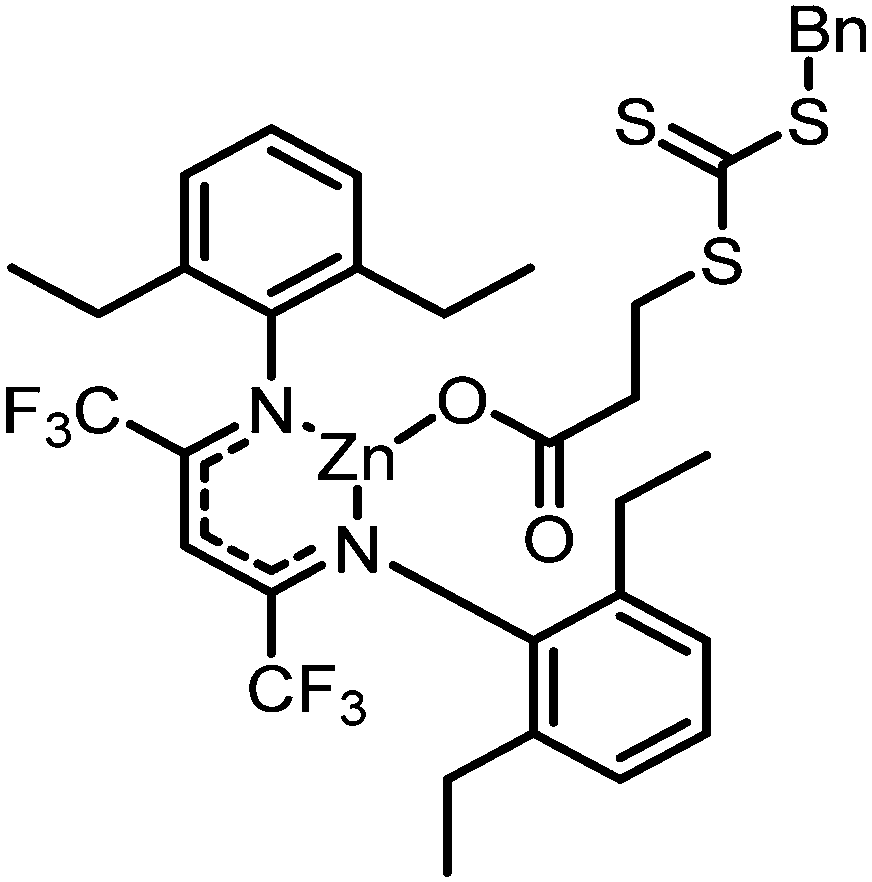
|
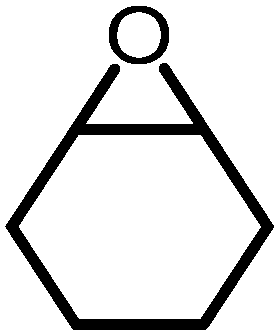
|
30 atm, 50 °C, 3 h, 1 mL toluene | M n = 28.6 kDa polymer, PDI = 1.11, Tg = 112 °C, block-polymer achieved with addition of N-isopropylacrylamide | 94, 2018 |
| 4 |
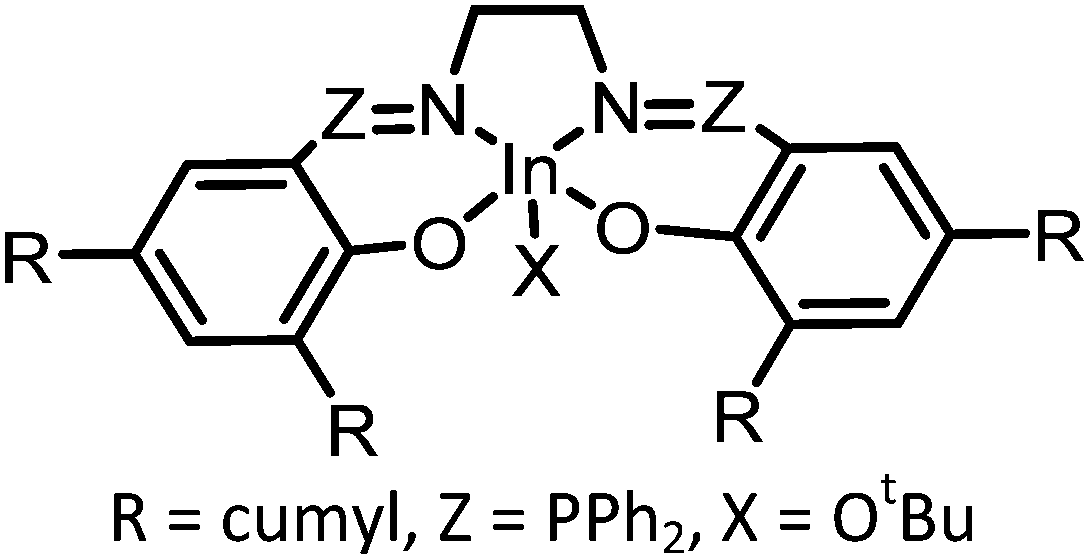
|

|
1 atm, 80 °C, 23 h, 0.1 mol% | >99% carbonate linkage, TON = 350, Mn = 3400 Da | 98, 2018 |
The increasing importance of CO2-epoxide derived pCs has also led to their application in other types of polymerization reactions. For example, living reversible addition–fragmentation chain transfer (RAFT) has been applied to the reaction, which leads to polymers with very narrow PDIs (<1.11). RAFT polymerization has been achieved using a β-diiminate Zn catalyst (see Fig. 4, the structure is structurally related to that of catalyst 8 in Fig. 3).94 Living polymerization methods also allow more complex block co-polymers to be prepared. In this respect, CO2-epoxide ring-opening copolymerization (ROCOP) has been combined with RAFT polymerization of vinyl monomers.103 Achieving controlled polymerization and block co-polymerization could, in turn, lead to polymers with superior properties, since pure pCs based on PC or limonene carbonate suffer from low rigidity and brittleness, respectively.79 Furan-containing monomers have been prepared and converted into their corresponding pC. This is particularly relevant in terms of sustainability because the furan ring is present in 5-hydroxymethylfurfural (5-HMF), a platform chemical obtained from cellulose (see Fig. 9, right).104 A useful feature of furan rings is that they react via well-established Diels–Alder (DA) chemistry (see Fig. 4 for an example), which enables post-polymerization functionalization.104–106 In the example given in Fig. 4 (top), the polymer is reacted with 10 equivalents of maleimide for 48 h at 60 °C (maleimides may be obtained from renewable resources). A retro-DA reaction proceeds at 100 °C and restores the furan-containing polymer. Interestingly, if the polymer contains both an olefin and a furan ring, reversible cross-linking of the polymer can be achieved using the DA reaction.107 Another reversible cross-linking strategy is to react the furan containing polymer with a bis-maleimide (Fig. 4, bottom).106,108 In the example shown, however, only 50% of the native polymer could be recovered following the retro-DA reaction. Moreover, the reaction requires a large excess of the dienophile when no catalyst is employed (10 eq. for the example in Fig. 4, top).109
 | ||
| Fig. 4 Schematic representation of post-polymerization functionalization of furan-containing pC using the DA reaction. | ||
Table 2 compiles the reaction conditions required by selected catalysts to copolymerize CO2 and epoxides. As for COC synthesis discussed above, no straightforward analysis is possible due to the vastly different reaction conditions employed. However, salen ligands are privileged ligands for the copolymerization reaction (Table 2, entries 2 and 4, as well as catalysts 7 and 9, Fig. 3). In most cases, an ionic/basic co-catalyst is required, and in some cases is covalently linked to the ligand (for example in catalyst 7, Fig. 3). Typically, the co-polymerization reaction is conducted at low temperatures and pressures (<100 °C, <30 atm, Table 2, entries 1–4). The resulting polymers often display a high Mn (for example 27 kDa, Table 2, entry 2) and narrow PDIs (<1.2). Notably, the Zn catalyst depicted in Table 2, entry 3, affords high Mn (28.6 kDa) homodispersed polymers (PDI = 1.11) at 50 °C and a moderate CO2 pressure (30 atm). The examples discussed above were presented without taking into account that the polymerization leads to two different stereoisomeric products as the epoxide starting material possesses a chiral center. Advantageously, catalysts have been developed that lead to a single, highly crystalline isotactic product. Producing isotactic polymers confers different properties to a material compared to a material displaying low or no tacticity, and highlights the crucial role of the catalysis in the polymerization reaction.110,111 Obtaining fully regio- and stereo-controlled polymers usually also leads to higher glass transition temperatures.92 A generic example of isotactic polycarbonate is shown in Fig. 5. Control over the stereoselectivity requires an appropriate catalyst, although the substituents on the epoxide can play a role as well. For instance, using a Co(III) catalyst, it has been shown that an electron withdrawing group on the epoxide can lead to highly isotactic polymers, allowing semi-crystalline and crystalline polymers to be obtained.112
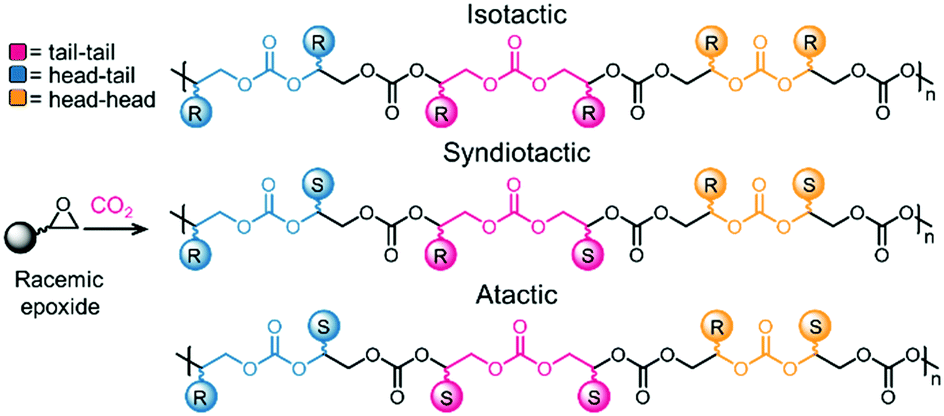 | ||
| Fig. 5 Example of stereoselective control during pC synthesis. Top: All-R stereocenter. Middle: Alternating R–S stereocenters. Bottom: Random R–S distribution.111 | ||
PHU synthesis from COCs and diamines
Definition of PUs and conventional synthesis methods
As mentioned in the Introduction, the definition of PUs is not restricted to polymers containing only urethane linkages. Rather, many classes of polymers are referred to as PUs because they contain urethane groups. Block polymers containing polyethers, polyesters or silicone polymers, in addition to PU segments, may be classified as PUs.114 Consequently, PUs have a large number of applications. PUs typically contain both rigid and flexible parts, and it is this combination that confers their thermoplastic properties (see Fig. 6).115,116 The chemistry of PUs has been intensively studied and numerous reviews and books describe the properties and applications of PUs.26,115,117–119 Similarly to PUs, PHU elastomers comprising rigid and flexible segments can be conveniently obtained from the COC-diamine route described below.120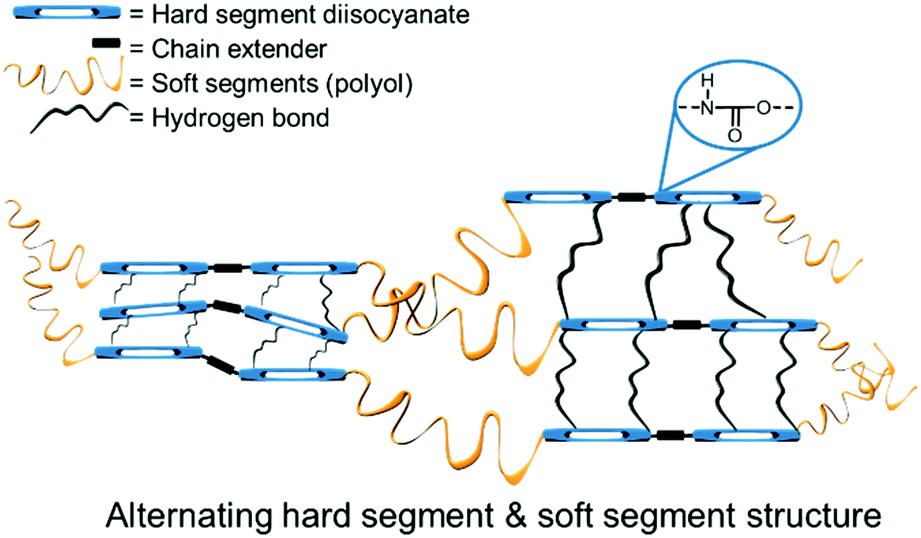 | ||
| Fig. 6 Schematic structure of a typical PU.119 | ||
The most common route to prepare PUs is via the reaction of isocyanates with diols (Fig. 7).121 The isocyanate functional group is water sensitive and the reaction between isocyanate and water leads to the release of CO2. For processing of PUs into foam-like structures, water is added intentionally to the process,122 but this method requires using an excess of isocyanate and may be less economically viable than other methods.123 The addition of water leads to a decrease in the density of the polymer, which is an important parameter for foams. PUs are routinely prepared on an industrial scale, and the materials find applications as sponges, in foams for car seats, in mattresses, and even in medicine.79,123Fig. 7 shows selected monomers (isocyanates, diols and diamines) and catalysts that are routinely employed in the synthesis of PUs.26 In general, N-containing bases are used as catalysts (e.g. DABCO, BDMAEE, etc., see Fig. 7).124 Notably, the reaction produces a single product and, unlike pC synthesis, a chiral catalyst is not needed for the reaction.
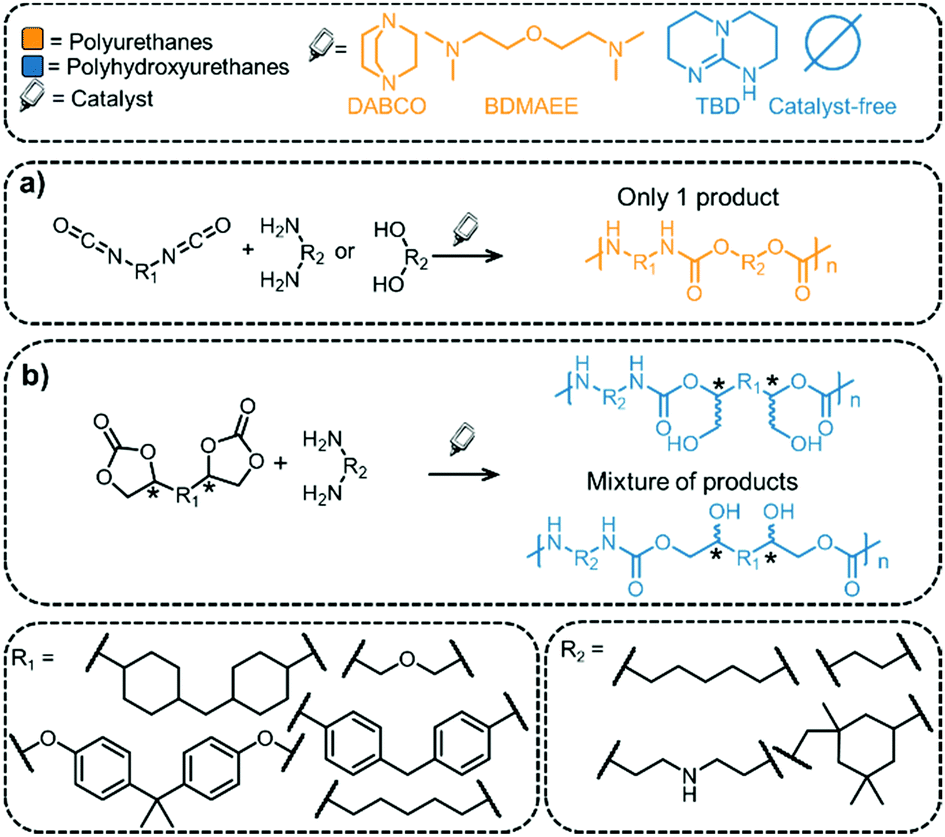 | ||
| Fig. 7 Synthesis of PUs and PHUs and selected functional groups and catalysts that have been used. | ||
PHU synthesis from COCs and diamines and their applications
In contrast to PUs, PHUs (also referred to as “non-isocyanate polyurethanes”, NIPUs) prepared from COCs and diamines have been less well investigated, but several excellent contributions demonstrate their high potential.125–129 Similar to PUs, PHUs derived from COCs and diamines have been prepared by combining different COCs and amine precursors, some examples of which are illustrated in Fig. 7.130 Examples of COCs employed include bisphenol A diglycydyl ether carbonate (BPAC) and diglycerol dicarbonate (DGC); the former is not derived from biomass sources but has been used routinely in pC packaging for decades. Unfortunately, BPA is harmful (it is being banned in food packaging), and alternatives are being investigated,131,132 for example DGC, a presumably less toxic molecule that can be derived from (renewable) glycerol.133Any diol can be reacted with ECH to generate bis(epoxides) that can be converted to the corresponding COCs.134 The diols can be derived from biomass sources (for example cellulose or lignin, see Fig. 8 for examples).135–137 Biomass conversion can lead to a variety of functional groups including acids, ketones, and others (alcohols, amines).138 Acids and ketones can be converted to alcohols by hydrogenation, and subsequently to COCs using ECH and the CCE reaction presented above. Note that acids or ketones obtained from biomass can also be used as platform chemicals for other polymeric products, such as poly lactic acid (pLA, one of the most important bio-based polymer that is biodegradable) or poly γ-valerolactone (pGVL).139 Diols can be obtained from lignin containing biomass sources by catalytic reductive fractionation with hydrogen and typically employ supported metal catalyst (Pd, Ni, Ru, Rh). Fig. 8 contains examples of structures of diols that may be obtained from biomass sources.140,141
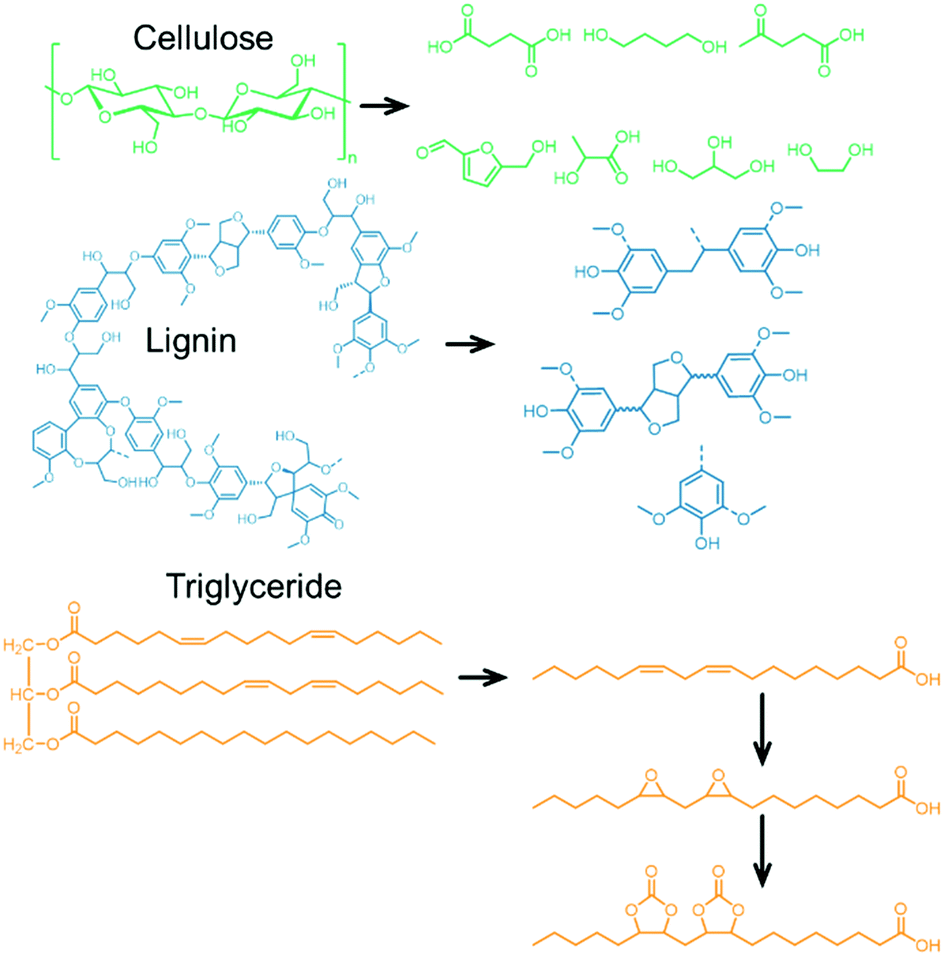 | ||
| Fig. 8 Examples of alcohols/diols derived from cellulose and lignin (top, middle) after hydrogenation/hydrogenolysis reaction. Example of a carbonated vegetable oil (linoleic acid, bottom) obtained from a natural triglyceride. | ||
1,2-Diols obtained from carbohydrate sources are converted to COCs using the highly toxic phosgene route or dimethyl carbonate (transesterification reaction).142 Notably, 1,2-diols can also be converted to COCs using CO2 as the C1 source, although this reaction is hindered by the release of water as a by-product (thermodynamic limitation, the reverse reaction is employed to generate EG from EC and water),143,144 which leads to moderate yields and significant amounts of waste because of the water capture strategies (utilization of alkyl halides as dehydrating agents, for example). COCs can also be obtained from vegetable oils or fatty acids because they contain olefin bonds. The olefins are first epoxidized and further transformed into their corresponding COC (see Fig. 8, bottom).122 These oil-based COCs can then be converted to a PHU by reaction with a suitable amine.145,146 This strategy is attractive because of the availability of the vegetable oil starting material and the properties of the resulting material that can be used as coatings and thermal insulators, etc.122,147
The dehydration of ethanol (obtained from cellulosic feedstock) yields ethylene, which can be oxidized to afford ethylene oxide,148 a substrate in the CCE reaction. Sugar-based polymers have attracted attention due to the availability of non-edible cellulosic feedstocks (which can be broken down into sugars), and examples of the polymerization of glucose and related compounds have been reviewed elsewhere.149,150 The hydrolysis/dehydration of cellulose leads to carbohydrates that can be further converted into 5-hydroxymethylfurfural (5-HMF, see Fig. 9, right),151 that can be transformed into a bis-epoxide and subsequently converted into a bis(carbonate).72,152,153 To make the material fully renewable, the coupling partner to the COC must also be derived from renewable sources.154 The formation of bio-based diamines is also possible from lignin derivatives when ammonia is added into the reaction mixture as a reagent.155,156 Currently, ammonia is generated from the Haber–Bosch (HB) process, but efforts to produce more sustainable ammonia are underway.157 The synthesis of ammonia (Haber–Bosch) is energy-intensive and, therefore, the biopolymers chitin and chitosan are being investigated as alternative sources of nitrogen containing compounds. Chitin is the second most abundant biomass source after cellulose.154,158 Efforts to prepare bio-based amines from chitin have been reviewed previously.155,159Fig. 10 shows a generic strategy to obtain COCs and diamines from biomass sources and catalysts that have been employed in the synthesis of PHUs.
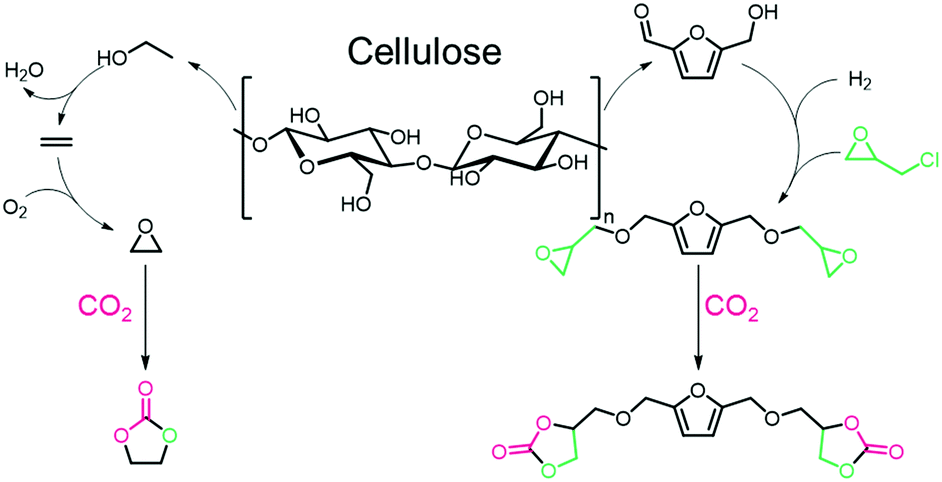 | ||
| Fig. 9 Preparation of renewable EC (left) and 5-HMF derived bis(carbonate) (right) from cellulose. | ||
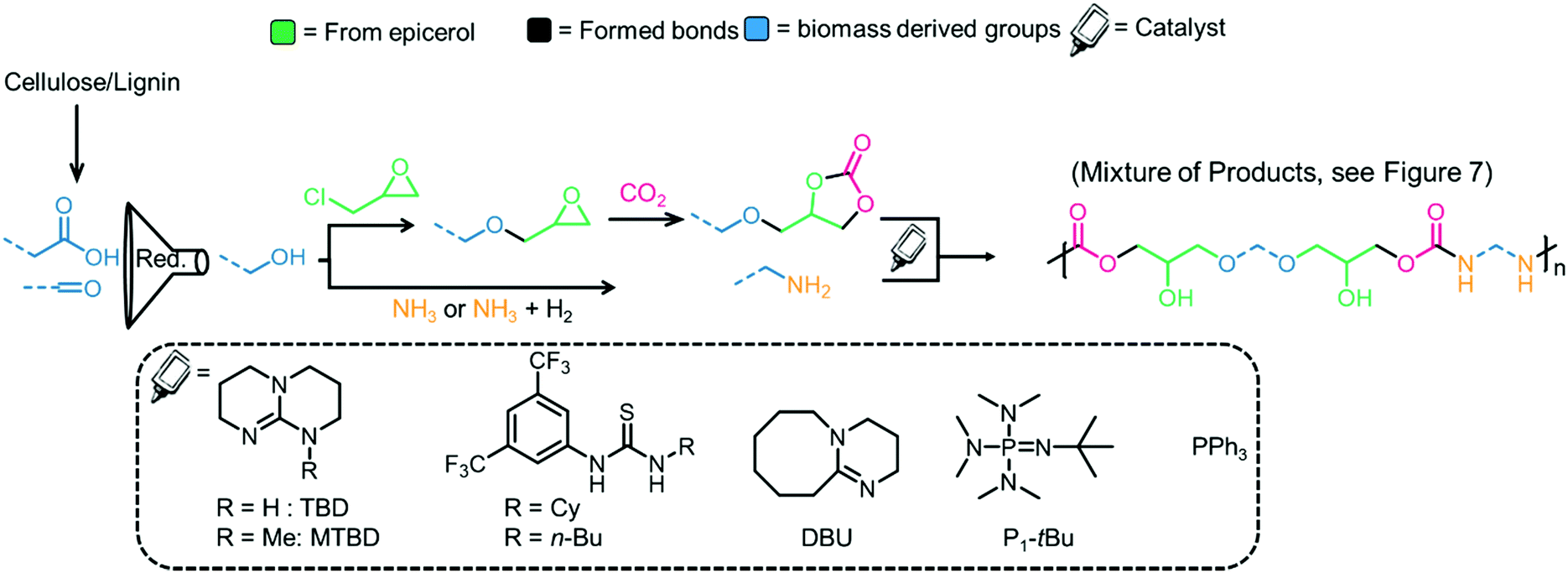 | ||
| Fig. 10 Generic strategy for the preparation of renewable PHUs from biomass sources and catalysts that have been used for PHU synthesis. The product is the result of the reaction between a bis-carbonate and a diamine. Only one side is presented for clarity.135,162,163 | ||
An example of a renewable PHU is depicted in Fig. 11, bottom, where the diamine is derived from the 5-HMF platform (furfural) by reaction with NH3 and the COC is derived from lignin.135 The polymer obtained from the compounds depicted in Fig. 11 is prepared in the molten phase, in the absence of a catalyst at 160 °C, and has a molecular weight of 5.3 kDa and a high PDI of 3.2. The polymer exhibits a glass transition temperature of 73 °C and decomposes at 267 °C, comparable to that of a bisphenol A polymer prepared under the same conditions (Tg = 79 °C and Td = 276 °C). PHUs obtained from isosorbides and Jeffamine (a polyetheramine based on propylene oxide/ethylene oxide) display properties suitable for coatings (low glass transition temperature of −8 to 59 °C).160 PHU vitrimers (polymers networks whose cross-links undergo associative exchange at elevated temperatures) have been prepared from COCs and diamines that possess similar mechanical properties to PU thermosets, but with enhanced reshaping and repair properties by compression molding (Fig. 11, top).129 The PHUs prepared from trimethylolpropane tris-carbonate (TMPTC) and commercial diamines such as 1,3-cyclohexanebis(methylamine) (CBMA) are used as adhesives and effectively bind to wood, aluminum and glass supports (Fig. 11, middle).161
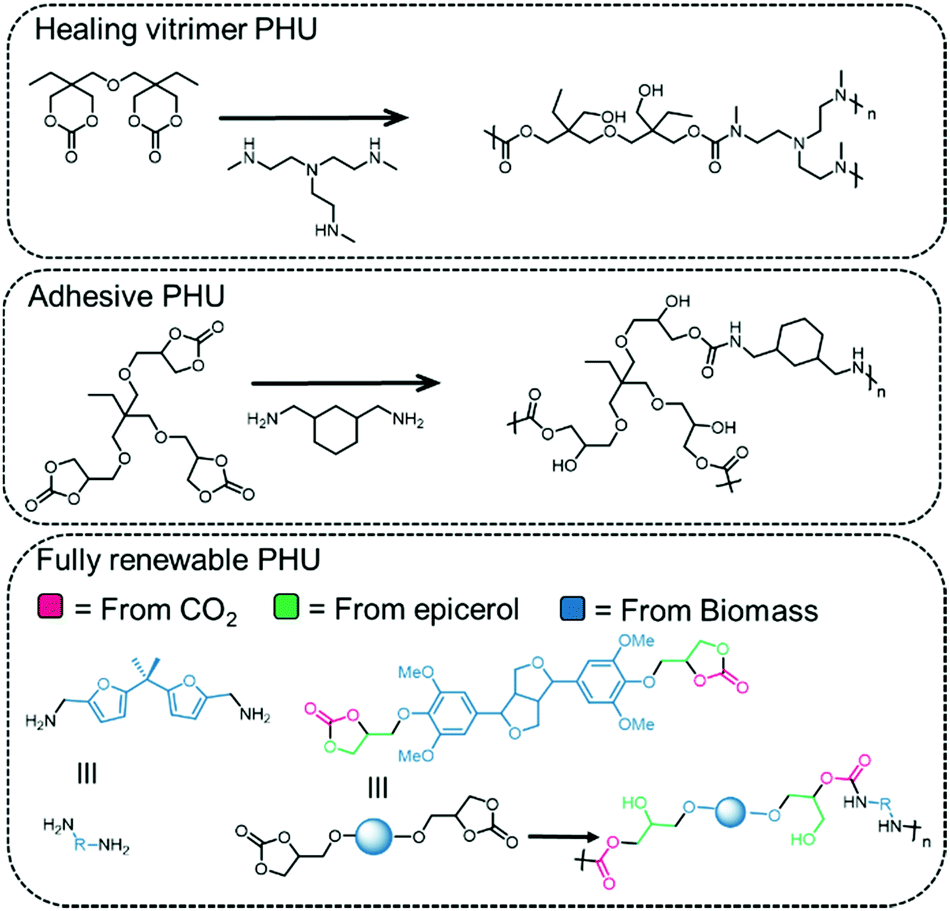 | ||
| Fig. 11 Examples of PHUs and their applications. | ||
Challenges in regiocontrol of PHUs
The structural difference between PUs and PHUs (i.e. one pendant alcohol group) confers very different properties to the polymers (more extensive H-bonding in the latter usually leads to higher glass transition temperatures, for example).164 It should be noted that it is difficult to control the regioselectivity during PHU synthesis and that often several different polymers (that differ in their regioselectivity or stereoselectivity) are formed (see Fig. 7).165 Chiral centers are also present in COCs and in the polymeric materials derived from them and a lack of stereocontrol leads to poorly defined polymers, which explains why PHUs are generally amorphous.127 PHUs typically have low molecular weights, which is attributed to the many side reactions that can occur during their synthesis, especially at higher temperatures. However, the presence of a pendant hydroxyl group in PHUs enables post-polymerization modifications.166,167 The side reactions that hinder high polymerization degrees of PHUs include the formation of ureas168 and oxazolidinones,20 (see Fig. 12 for representative side-reaction pathways that can occur in catalyzed and non-catalyzed reactions).127 At the same time, it is the relatively high reactivity of the urethane C–O bond that renders these polymers biodegradable.114,169 Foaming of PHUs is also problematic, since no gas is released during the synthesis (addition of water to isocyanate-based PUs forms CO2 – the foaming agent) which makes processing more challenging.122 Nevertheless, foaming strategies for non-isocyanate PUs have been proposed, using, for example, poly(methylhydrogenosiloxane) as a blowing agent.170,171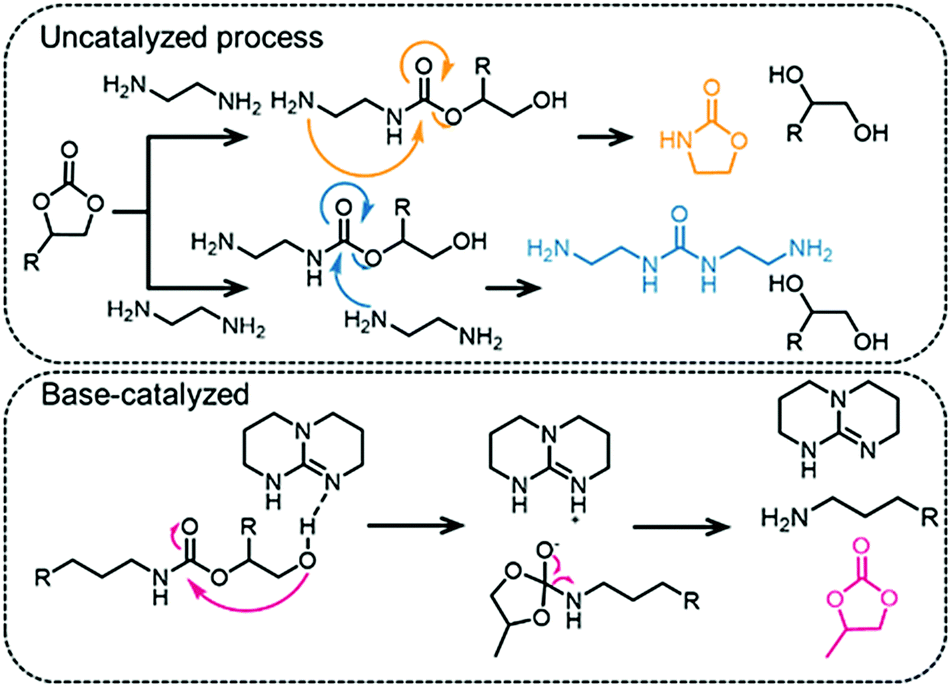 | ||
| Fig. 12 Unwanted side-reactions that take place during PHU synthesis from COCs and ethylenediamine. | ||
The catalysts used to prepare PHUs comprise a few N-bases, metal salts and thiourea compounds (see Fig. 10, examples include TBD and DABCO).162,165,172–174 While catalysts accelerate the reaction they currently do not solve the regioselectivity issues. Moreover, it was shown that if the catalyst is highly basic (for example TBD), then the selectivity can decrease and the reaction can lead to the formation of urea or COC functional groups rather than urethane linkages (see Fig. 12, bottom, for the mechanism of the TBD-catalyzed transformation).175
Strategies that lead to regioregular polymers are being investigated. It appears that one promising approach to obtain such polymers is to use/design suitable monomers. For example, using COCs and amines with bulky substituents leads to regioselective polymers when TBD is employed as a catalyst.176 Furthermore, when α-alkylidenes are used as the starting materials, regioregular PHUs are obtained (Fig. 13, top).177 Note that if two equivalents of EC is reacted with a diamine, then it is possible to avoid the pendant hydroxyl group and hence the regioselectivity issues. The resulting product is a diurethane diol that can be transformed into polyesters–polyurethanes (Fig. 13, bottom).178 Recently, trans-carbamates were obtained by taking advantage of existing tools for pC synthesis (see above). Incorporating an amine into an epoxide:CO2:catalyst mixture leads to trans-carbamate products in the presence of chiral aluminum or cobalt catalysts (Fig. 14).179,180 Presumably, this strategy could open a way to controlled polymerization by reacting the epoxide:CO2 mixture with diamines.
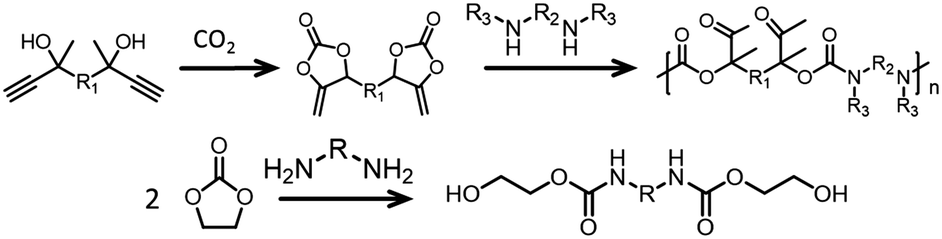 | ||
| Fig. 13 Example of regioregular poly(oxourethane) (top) and diurethane diol (bottom).177 | ||
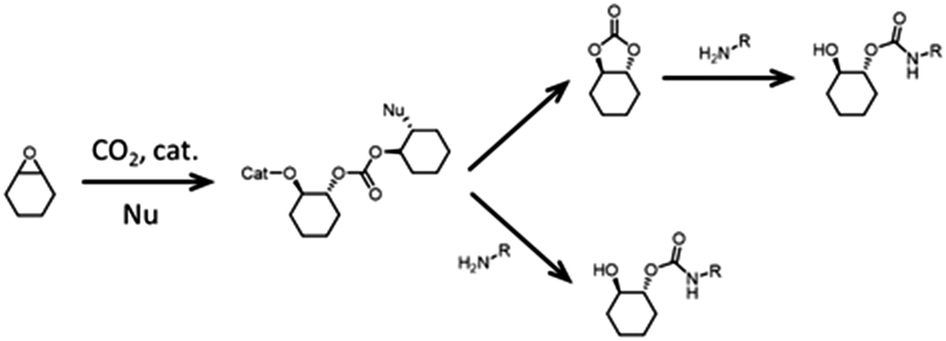 | ||
| Fig. 14 Trapping of a trans-carbamate product via a pC intermediate. | ||
Biodegradation of the polymers/polymer end-of-life
It is not only important that future polymers are based on renewable sources, but that they do not pose long-term environmental problems. Therefore, designing tomorrow's materials will require a better understanding of (bio)degradation pathways and how to ensure polymers degrade within reasonable time scales and do not release toxic by-products into the environment. Biodegradation requires chemical bonds to be cleaved by enzymes (present in microorganisms). Other degradation pathways such as oxidation, hydrolysis or irradiation are often considered as biodegradation, even if these pathways do not require enzymes/microorganisms.80 The behavior and degradation of polymers under physiological conditions, for example, has been extensively studied in the context of implants.181–183 Biodegradation often relies on a depolymerization step followed by a mineralization step. The first step converts the polymer into its monomer, and in the second step digestion of the monomer into (ideally) CO2 and water, takes place.184 Biodegradation is often limited as polymers cannot be readily accessed by the enzyme capable of cleaving the polymer bonds. Hence, polymers are required that can penetrate within the active site of enzymes. For instance, not all pCs are necessarily biodegradable and care must be taken when referring to pCs as biodegradable. In this respect, the pC based on bisphenol A is not readily degradable and persists in the environment.80Fig. 15 depicts a conceptual representation of enzymatic biodegradation. Biodegradation pathways are complex and a simple change in the polymeric structure can lead to major changes in the decomposition pathway. For example, polyethylene glycols (PEGs) and polypropylene glycols (PPGs) decompose via different routes.185
 | ||
| Fig. 15 Representation of enzyme catalyzed polymer degradation.80 | ||
The mechanism of biodegradation of PUs has been studied and several pathways are possible including oxidative biodegradation. If the PU contains polyesters or polyureas, then esterases or ureases are able to cleave the bonds and degrade the polymer. The prerequisite is that the polymer can reach the active site of the enzyme.186 In fact, including PUs in the structure of functional materials represents a strategy to increase the degradability of polymers that would otherwise not degrade. This strategy is employed in the preparation of biodegradable conducting polymers for biomedical applications.187,188 Notably, even if the materials are bio-derived, there is no guarantee that the resulting polymer will be degradable (for example if the material is highly cross-linked and hydrophobic).189 Thermal degradation pathways of the polymers can result in the release of toxic chemicals. The decomposition products from the thermal degradation of a PHU is depicted in Fig. 16.190 The main reactions include the reversible depolymerization reaction of the urethane bond to form an isocyanate and an alcohol. CO2 is also liberated upon degradation of the polymer, as well as amines and olefins.
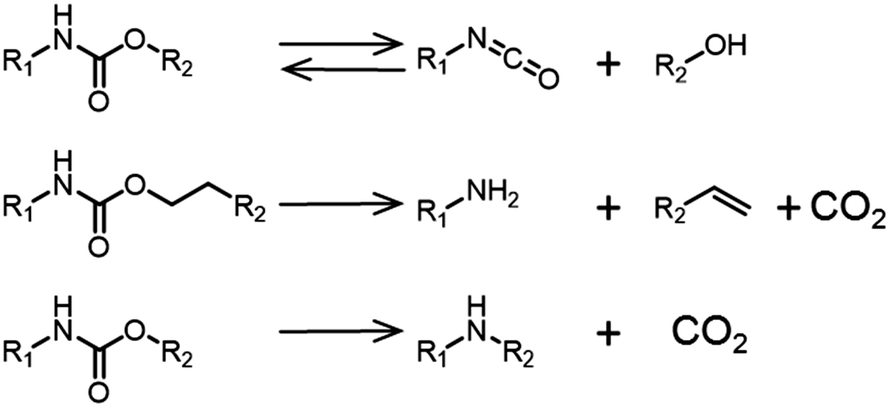 | ||
| Fig. 16 Decomposition pathways of urethane linkages at 200–300 °C.190 | ||
Chemically converting polymers into their monomers in order to reuse the monomers is an attractive recycling concept. This approach has been extensively studied for polyesters and pCs, where the polymeric linkages (esters or carbonates) can be disrupted by hydrolysis,191 alcoholysis,192 glycolysis193 or hydrogenolysis (see Fig. 17 that illustrates the chemical pathways for bisphenol carbonate depolymerization).194 Currently, it is more cost-effective to dispose of the waste and generate new polymers from petrochemicals than to recycle and reuse existing materials.195
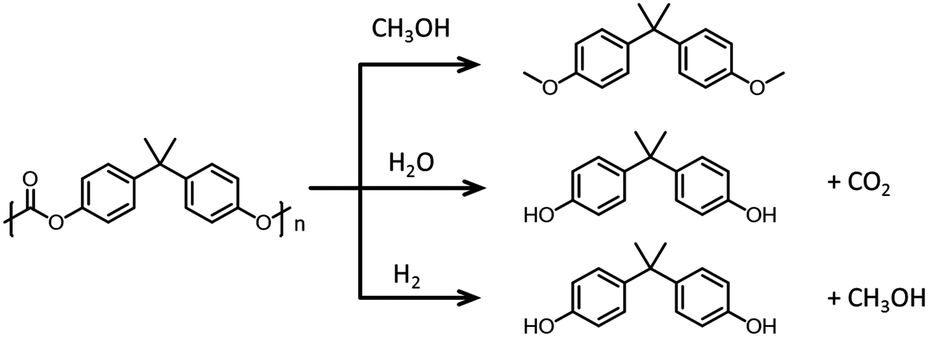 | ||
| Fig. 17 Chemical depolymerization of bisphenol A carbonate. Top: Methanolysis. Middle: Hydrolysis. Bottom: Hydrogenolysis. | ||
Current limitations, future perspectives and conclusions
The preparation of COCs has been extensively studied due to the wide range of uses of liquid COCs such as PC and EC. COCs are also used as reactive precursors to PUs and PCs. Terminal epoxides react smoothly in the presence of easily accessed catalysts and does not require elevated CO2 pressures, high temperatures or prolonged reaction times. Indeed, the reaction can be conducted under diluted, continuous-flow reaction conditions at atmospheric pressure.196,197 Hence, thermally robust catalysts that are resistant to water, oxygen, and industrial contaminants such as H2S are sought after.Suitable terminal epoxides can be derived from waste biomass sources. Importantly, for the process to be sustainable the use of waste (inedible) biomass is essential rather than using biomass from crops specifically grown to produce a chemical (non-food) product.
The reaction between COCs and diamines (or aminols/diols) leads to PHUs (or pCs), and it seems likely that bio-derived PHUs will find large-scale applications in the future. Although PHUs are prepared from COCs and diamines in the absence of a catalyst, discovering catalysts that enable a higher degree of regio- and stereo-control should afford polymers with more homogeneous properties and will accelerate the utilization of renewable PHUs at an industrial level.
The catalysts described herein are derived from petrochemicals. The catalysts are not used in the same quantities as the chemicals they are used to produce, but if the preparation of renewable materials must become fully sustainable, the development of renewable catalysts becomes relevant. In this respect, catalysts based on lecithin,198,199 chitosan200 or animal-based wool have been used for COC synthesis100 and natural catalysts/supports are being exploited in other applications.201,202
Finally, a thorough life-cycle analysis (LCA) of the obtained polymers is required to assess the sustainability of the process. For example, CO2-containing materials do not necessarily decrease the net amount of CO2 since energy is required for their transformations.203 Further, at the “end-of-life”, the polymers will typically be converted into CO2 again, as mentioned in the section on biodegradation. An extensive review on the LCA of CO2-derived chemicals has been published very recently and these types of analysis are extremely relevant to pursue the quest of catalytically produced sustainable materials.204
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the EPFL, the CTI Swiss Competence Center for Energy Research (SCCER) on Heat and Electricity Storage, and the Swiss National Science Foundation for financial support.Notes and references
- A. W. Kleij, M. North and A. Urakawa, ChemSusChem, 2017, 10, 1036–1038 CrossRef CAS PubMed.
- S. A. Zimov, E. A. G. Schuur and F. S. Chapin III, Science, 2006, 312, 1612–1613 CrossRef CAS PubMed.
- C. Le Quéré, R. Moriarty, R. M. Andrew, G. P. Peters, P. Ciais, P. Friedlingstein, S. D. Jones, S. Sitch, P. Tans, A. Arneth, T. A. Boden, L. Bopp, Y. Bozec, J. G. Canadell, L. P. Chini, F. Chevallier, C. E. Cosca, I. Harris, M. Hoppema, R. A. Houghton, J. I. House, A. K. Jain, T. Johannessen, E. Kato, R. F. Keeling, V. Kitidis, K. Klein Goldewijk, C. Koven, C. S. Landa, P. Landschützer, A. Lenton, I. D. Lima, G. Marland, J. T. Mathis, N. Metzl, Y. Nojiri, A. Olsen, T. Ono, S. Peng, W. Peters, B. Pfeil, B. Poulter, M. R. Raupach, P. Regnier, C. Rödenbeck, S. Saito, J. E. Salisbury, U. Schuster, J. Schwinger, R. Séférian, J. Segschneider, T. Steinhoff, B. D. Stocker, A. J. Sutton, T. Takahashi, B. Tilbrook, G. R. Van Der Werf, N. Viovy, Y. P. Wang, R. Wanninkhof, A. Wiltshire and N. Zeng, Earth Syst. Sci. Data, 2015, 7, 47–85 CrossRef.
- P. Falkowski, Science, 2013, 291, 290–296 Search PubMed.
- T. P. Hughes, J. T. Kerry, M. Álvarez-Noriega, J. G. Álvarez-Romero, K. D. Anderson, A. H. Baird, R. C. Babcock, M. Beger, D. R. Bellwood, R. Berkelmans, T. C. Bridge, I. R. Butler, M. Byrne, N. E. Cantin, S. Comeau, S. R. Connolly, G. S. Cumming, S. J. Dalton, G. Diaz-Pulido, C. M. Eakin, W. F. Figueira, J. P. Gilmour, H. B. Harrison, S. F. Heron, A. S. Hoey, J. A. Hobbs, M. O. Hoogenboom, E. V. Kennedy, C. Kuo, J. M. Lough, R. J. Lowe, G. Liu, M. T. McCulloch, H. A. Malcolm, M. J. McWilliam, J. M. Pandolfi, R. J. Pears, M. S. Pratchett, V. Schoepf, T. Simpson, W. J. Skirving, B. Sommer, G. Torda, D. R. Wachenfeld, B. L. Willis and S. K. Wilson, Nature, 2017, 543, 373–377 CrossRef CAS PubMed.
- C. McGlade and P. Ekins, Nature, 2015, 517, 187–190 CrossRef CAS PubMed.
- B. Smit, A.-H. A. Park and G. Gadikota, Front. Energy Res., 2014, 2, 2013–2015 Search PubMed.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
- B. Walsh, Energy Environ. Sustain., 2013, 325, 1652–1655 Search PubMed.
- D. M. D’Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- R. Socolow, M. Desmond, R. Aines, J. Blackstock, O. Bolland, T. Kaarsberg, N. Lewis, M. Mazzotti, A. Pfeffer, K. Sawyer, J. Siirola, B. Smit and J. Wilcox, Am. Phys. Soc., 2011, 100 Search PubMed.
- N. R. Stuckert and R. T. Yang, Environ. Sci. Technol., 2011, 45, 10257–10264 CrossRef CAS PubMed.
- V. Favero and L. Laloui, Int. J. Greenhouse Gas Control, 2018, 71, 133–141 CrossRef CAS.
- I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef CAS.
- J. Meessen, Chem. Ing. Tech., 2014, 86, 2180–2189 CrossRef CAS.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed.
- B. Schäffner, S. P. Verevkin and A. Börner, Chem. Unserer Zeit, 2009, 43, 12–21 CrossRef.
- J. Peng and Y. Deng, New J. Chem., 2001, 25, 639–641 RSC.
- J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS.
- T. Yamada and H. Ono, Bioresour. Technol., 1999, 70, 61–67 CrossRef CAS.
- M. O. Sonnati, S. Amigoni, E. P. Taffin de Givenchy, T. Darmanin, O. Choulet and F. Guittard, Green Chem., 2013, 15, 283–306 RSC.
- Huntsman Corporation, Tech. Data Sheet, 2011, 2509.
- S. Sopeña, G. Fiorani, C. Martín and A. W. Kleij, ChemSusChem, 2015, 8, 3248–3254 CrossRef PubMed.
- C. J. Whiteoak, A. H. Henseler, C. Ayats, A. W. Kleij and M. A. Pericàs, Green Chem., 2014, 16, 1552–1559 RSC.
- A. L. Silva and J. C. Bordado, Catal. Rev.: Sci. Eng., 2004, 46, 31–51 CrossRef.
- G. Wegener, M. Brandt, L. Duda, J. Hofmann, B. Klesczewski, D. Koch, R. J. Kumpf, H. Orzesek, H. G. Pirkl, C. Six, C. Steinlein and M. Weisbeck, Appl. Catal., A, 2001, 221, 303–335 CrossRef CAS.
- R. B. Seymour and G. B. Kauffman, J. Chem. Educ., 1992, 69, 909–910 CrossRef CAS.
- C. Six and F. Richter, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2003, pp. 264–322 Search PubMed.
- J. R. Bucher, Toxicol. Sci, 1987, 9, 367–379 CrossRef CAS.
- F. Zia, K. M. Zia, M. Zuber, S. Kamal and N. Aslam, Carbohydr. Polym., 2015, 134, 784–798 CrossRef CAS PubMed.
- A. Usman, K. M. Zia, M. Zuber, S. Tabasum, S. Rehman and F. Zia, Int. J. Biol. Macromol., 2016, 86, 630–645 CrossRef CAS PubMed.
- A. Noreen, K. M. Zia, M. Zuber, S. Tabasum and A. F. Zahoor, Prog. Org. Coat., 2016, 91, 25–32 CrossRef CAS.
- N. Mahmood, Z. Yuan, J. Schmidt and C. Xu, Renewable Sustainable Energy Rev., 2016, 60, 317–329 CrossRef CAS.
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- Y. Li, X. Cui, K. Dong, K. Junge and M. Beller, ACS Catal., 2017, 7, 1077–1086 CrossRef CAS.
- M. H. Beyzavi, C. J. Stephenson, Y. Liu, O. Karagiaridi, J. T. Hupp and O. K. Farha, Front. Energy Res., 2015, 2, 1–10 Search PubMed.
- M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed.
- T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J. Y. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270–15279 CrossRef CAS PubMed.
- C. Miceli, J. Rintjema, E. Martin, E. C. Escudero-Adán, C. Zonta, G. Licini and A. W. Kleij, ACS Catal., 2017, 7, 2367–2373 CrossRef CAS.
- T. Sakai, N. Kihara and T. Endo, Macromolecules, 1995, 28, 4701–4706 CrossRef CAS.
- V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef.
- T. Iwasaki, N. Kihara and T. Endo, Bull. Chem. Soc. Jpn., 2000, 73, 713–719 CrossRef CAS.
- W. Zhong, F. D. Bobbink, Z. Fei and P. J. Dyson, ChemSusChem, 2017, 10, 2728–2735 CrossRef CAS PubMed.
- F. D. Bobbink, A. P. Van Muyden, A. Gopakumar, Z. Fei and P. J. Dyson, ChemPlusChem, 2017, 82, 144–151 CrossRef CAS.
- F. D. Bobbink, Z. Fei, R. Scopelliti, S. Das, P. J. Dyson and J. Paul, Helv. Chim. Acta, 2016, 99, 821–829 CrossRef CAS.
- S. Ghazali-Esfahani, H. Song, E. Păunescu, F. D. Bobbink, H. Liu, Z. Fei, G. Laurenczy, M. Bagherzadeh, N. Yan and P. J. Dyson, Green Chem., 2013, 15, 1584–1589 RSC.
- T. P. Thuy Pham, C. W. Cho and Y. S. Yun, Water Res., 2010, 44, 352–372 CrossRef PubMed.
- D. Coleman and N. Gathergood, Chem. Soc. Rev., 2010, 39, 600–637 RSC.
- A. Jordan and N. Gathergood, Chem. Soc. Rev., 2015, 44, 8200–8237 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- K. R. Roshan, T. Jose, D. Kim, K. A. Cherian and D. W. Park, Catal. Sci. Technol., 2014, 4, 963–970 RSC.
- K. Yamaguchi, K. Ebitani, T. Yoshida, H. Yoshida and K. Kaneda, J. Am. Chem. Soc., 1999, 121, 4526–4527 CrossRef CAS.
- F. D. Bobbink and P. J. Dyson, J. Catal., 2016, 343, 52–61 CrossRef CAS.
- Q. He, J. W. O’Brien, K. A. Kitselman, L. E. Tompkins, G. C. T. Curtis and F. M. Kerton, Catal. Sci. Technol., 2014, 4, 1513–1528 RSC.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- F. D. Bobbink, D. Vasilyev, M. Hulla, S. Chamam, F. Menoud, G. Laurenczy, S. Katsyuba and P. J. Dyson, ACS Catal., 2018, 8, 2589–2594 CrossRef CAS.
- N. Kihara, N. Hara and T. Endo, J. Org. Chem., 1993, 58, 6198–6202 CrossRef CAS.
- S. Marmitt and P. F. B. Gonçalves, J. Comput. Chem., 2015, 36, 1322–1333 CrossRef CAS PubMed.
- S. Gennen, M. Alves, R. Méreau, T. Tassaing, B. Gilbert, C. Detrembleur, C. Jerome and B. Grignard, ChemSusChem, 2015, 8, 1845–1849 CrossRef CAS PubMed.
- G. M. Lari, G. Pastore, C. Mondelli and J. Pérez-Ramírez, Green Chem., 2018, 20, 148–159 RSC.
- W. H. Lawrence, M. Malik, J. E. Turner and J. Autian, J. Pharm. Sci., 1972, 61, 1712–1717 CrossRef CAS PubMed.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 4990–5011 RSC.
- J. Rintjema and A. W. Kleij, ChemSusChem, 2017, 10, 1274–1282 CrossRef CAS PubMed.
- M. Hong, Y. Kim, H. Kim, H. J. Cho, M. Baik and Y. Kim, J. Org. Chem., 2018, 83, 9370–9380 CrossRef CAS PubMed.
- J. Sun, J. Ren, S. Zhang and W. Cheng, Tetrahedron Lett., 2009, 50, 423–426 CrossRef CAS.
- P. Yan and H. Jing, Adv. Synth. Catal., 2009, 351, 1325–1332 CrossRef CAS.
- R. L. Paddock and S. T. Nguyen, Chem. Commun., 2004, 1622–1623 RSC.
- M. North, S. C. Z. Quek, N. E. Pridmore, A. C. Whitwood and X. Wu, ACS Catal., 2015, 5, 3398–3402 CrossRef CAS.
- X. B. Lu, B. Liang, Y. J. Zhang, Y. Z. Tian, Y. M. Wang, C. X. Bai, H. Wang and R. Zhang, J. Am. Chem. Soc., 2004, 126, 3732–3733 CrossRef CAS PubMed.
- J. Hu, J. Ma, H. Liu, Q. Qian, C. Xie and B. Han, Green Chem., 2018, 20, 2990 RSC.
- J. A. Castro-Osma, J. Martínez, F. De La Cruz-Martínez, M. P. Caballero, J. Fernández-Baeza, J. Rodríguez-López, A. Otero, A. Lara-Sánchez and J. Tejeda, Catal. Sci. Technol., 2018, 8, 1981–1987 RSC.
- L. Wang, G. Zhang, K. Kodama and T. Hirose, Green Chem., 2016, 18, 1229–1233 RSC.
- K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2006, 45, 7274–7277 CrossRef CAS PubMed.
- M. Scharfenberg, J. Hilf and H. Frey, Adv. Funct. Mater., 2018, 28, 1–16 CrossRef.
- Y. Qin and X. Wang, Biotechnol. J., 2010, 5, 1164–1180 CrossRef CAS PubMed.
- X. B. Lu and D. J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 RSC.
- M. Taherimehr and P. P. Pescarmona, J. Appl. Polym. Sci., 2014, 131, 1–17 CrossRef.
- T. Artham and M. Doble, Macromol. Biosci., 2008, 8, 14–24 CrossRef CAS PubMed.
- F. Auriemma, C. De Rosa, M. R. Di Caprio, R. Di Girolamo, W. C. Ellis and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 1215–1218 CrossRef CAS PubMed.
- O. Hauenstein, S. Agarwal and A. Greiner, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- E. H. Lee, J. Y. Ahn, M. M. Dharman, D. W. Park, S. W. Park and I. Kim, Catal. Today, 2008, 131, 130–134 CrossRef CAS.
- H. Sugimoto, H. Ohshima and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3549–3555 CrossRef CAS.
- O. Hauenstein, M. Reiter, S. Agarwal, B. Rieger and A. Greiner, Green Chem., 2016, 18, 760–770 RSC.
- L. S. Morris, M. I. Childers and G. W. Coates, Angew. Chem., Int. Ed., 2018, 57, 5731–5734 CrossRef CAS PubMed.
- M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. Lapointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- W. Hirahata, R. M. Thomas, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2008, 130, 17658–17659 CrossRef CAS PubMed.
- R. M. Thomas, P. C. B. Widger, S. M. Ahmed, R. C. Jeske, W. Hirahata, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2010, 132, 16520–16525 CrossRef CAS PubMed.
- M. I. Childers, A. K. Vitek, L. S. Morris, P. C. B. Widger, S. M. Ahmed, P. M. Zimmerman and G. W. Coates, J. Am. Chem. Soc., 2017, 139, 11048–11054 CrossRef CAS PubMed.
- K. N. Eun, J. N. Sung, S. Sujith, S. W. Kim and Y. L. Bun, J. Am. Chem. Soc., 2007, 129, 8082–8083 CrossRef PubMed.
- Y. Xu, L. Lin, M. Xiao, S. Wang, A. T. Smith, L. Sun and Y. Meng, Prog. Polym. Sci., 2018, 80, 163–182 CrossRef CAS.
- J. Y. Jeon, J. J. Lee, J. K. Varghese, S. J. Na, S. Sujith, M. J. Go, J. Lee, M.-A. Ok and B. Y. Lee, Dalton Trans., 2013, 42, 9245–9254 RSC.
- Y. Y. Zhang, G. W. Yang and G. P. Wu, Macromolecules, 2018, 51, 3640–3646 CrossRef CAS.
- R. K. Dean, K. Devaine-Pressing, L. N. Dawe and C. M. Kozak, Dalton Trans., 2013, 42, 9233–9244 RSC.
- J. Gurnham, S. Gambarotta, I. Korobkov, L. Jasinska-Walc and R. Duchateau, Organometallics, 2014, 33, 4401–4409 CrossRef CAS.
- M. Taherimehr, J. P. C. C. Sertã, A. W. Kleij, C. J. Whiteoak and P. P. Pescarmona, ChemSusChem, 2015, 8, 1034–1042 CrossRef CAS PubMed.
- A. Thevenon, A. Cyriac, D. Myers, A. J. P. White, C. B. Durr and C. K. Williams, J. Am. Chem. Soc., 2018, 140, 6893–6903 CrossRef CAS PubMed.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618–6639 CrossRef CAS PubMed.
- Y. L. Bun, Y. K. Heon, Y. L. Su, J. N. Sung, S. I. Han, H. Yun, H. Lee and Y. W. Park, J. Am. Chem. Soc., 2005, 127, 3031–3037 CrossRef PubMed.
- M. Cozzolino, K. Press, M. Mazzeo and M. Lamberti, ChemCatChem, 2016, 8, 455–460 CrossRef CAS.
- D. Zhang, S. K. Boopathi, N. Hadjichristidis, Y. Gnanou and X. Feng, J. Am. Chem. Soc., 2016, 138, 11117–11120 CrossRef CAS PubMed.
- Y. Wang, Y. Zhao, Y. Ye, H. Peng, X. Zhou, X. Xie, X. Wang and F. Wang, Angew. Chem., Int. Ed., 2018, 57, 3593–3597 CrossRef CAS PubMed.
- J. Hilf, M. Scharfenberg, J. Poon, C. Moers and H. Frey, Macromol. Rapid Commun., 2015, 36, 174–179 CrossRef CAS PubMed.
- A. Dag, M. Aydin, H. Durmaz, G. Hizal and U. Tunca, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4476–4483 CrossRef CAS.
- M. J. Barthel, T. Rudolph, S. Crotty, F. H. Schacher and U. S. Schubert, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4958–4965 CrossRef CAS.
- Z. Mou and E. Y. X. Chen, ACS Sustainable Chem. Eng., 2016, 4, 7118–7129 CrossRef CAS.
- E. Slavko and M. S. Taylor, Chem. Sci., 2017, 8, 7106–7111 RSC.
- V. Froidevaux, C. Negrell, S. Caillol, J. P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- M. Taherimehr, S. M. Al-Amsyar, C. J. Whiteoak, A. W. Kleij and P. P. Pescarmona, Green Chem., 2013, 15, 3083–3090 RSC.
- N. Kielland, C. J. Whiteoak and A. W. Kleij, Adv. Synth. Catal., 2013, 355, 2115–2138 CrossRef CAS.
- W. M. Ren, T. J. Yue, X. Zhang, G. G. Gu, Y. Liu and X. B. Lu, Macromolecules, 2017, 50, 7062–7069 CrossRef CAS.
- M. Hatazawa, K. Nakabayashi, S.-i Ohkoshi and K. Nozaki, Chem. – Eur. J., 2016, 22, 13677–13681 CrossRef CAS PubMed.
- E. M. Christenson, J. M. Anderson and A. Hiltner, Corros. Eng., Sci. Technol., 2007, 42, 312–323 CrossRef CAS.
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- A. M. Nelson and T. E. Long, Macromol. Chem. Phys., 2014, 215, 2161–2174 CrossRef CAS.
- X. Zhou, Y. Li, C. Fang, S. Li, Y. Cheng, W. Lei and X. Meng, J. Mater. Sci. Technol., 2015, 31, 708–722 CrossRef CAS.
- I. Yilgör, E. Yilgör and G. L. Wilkes, Polymer, 2015, 58, A1–A36 CrossRef.
- C. Prisacariu, Polyurethane Elastomers, Springer, 2011 Search PubMed.
- J. Nanclares, Z. S. Petrovic, I. Javni, M. Ionescu and F. Jaramillo, J. Appl. Polym. Sci., 2015, 132, 7–14 CrossRef.
- E. Delebecq, J. P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- B. Grignard, J. M. Thomassin, S. Gennen, L. Poussard, L. Bonnaud, J. M. Raquez, P. Dubois, M. P. Tran, C. B. Park, C. Jerome and C. Detrembleur, Green Chem., 2016, 18, 2206–2215 RSC.
- M. Thirumal, D. Khastgir, N. K. Singha, B. S. Manjunath and Y. P. Naik, J. Appl. Polym. Sci., 2008, 108, 1810–1817 CrossRef CAS.
- P. Król, Prog. Mater. Sci., 2007, 52, 915–1015 CrossRef.
- H. Tomita, F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3678–3685 CrossRef CAS.
- B. Nohra, L. Candy, J.-F. Blanco, C. Guerin, Y. Raoul and Z. Mouloungui, Macromolecules, 2013, 46, 3771–3792 CrossRef CAS.
- V. Besse, F. Camara, F. Méchin, E. Fleury, S. Caillol, J. P. Pascault and B. Boutevin, Eur. Polym. J., 2015, 71, 1–11 CrossRef CAS.
- E. K. Leitsch, G. Beniah, K. Liu, T. Lan, W. H. Heath, K. A. Scheidt and J. M. Torkelson, ACS Macro Lett., 2016, 5, 424–429 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- J. Ke, X. Li, F. Wang, S. Jiang, M. Kang, J. Wang, Q. Li and Z. Wang, RSC Adv., 2017, 7, 28841–28852 RSC.
- E. J. Hoekstra and C. Simoneau, Crit. Rev. Food Sci. Nutr., 2013, 53, 386–402 CrossRef CAS PubMed.
- M.-Y. Chen, M. Ike and M. Fujita, Environ. Toxicol., 2002, 17, 80–86 CrossRef CAS PubMed.
- J. L. J. Van Velthoven, L. Gootjes, D. S. Van Es, B. A. J. Noordover and J. Meuldijk, Eur. Polym. J., 2015, 70, 125–135 CrossRef CAS.
- F. Ng, G. Couture, C. Philippe, B. Boutevin and S. Caillol, Molecules, 2017, 22, 149 CrossRef PubMed.
- M. Janvier, P. H. Ducrot and F. Allais, ACS Sustainable Chem. Eng., 2017, 5, 8648–8656 CrossRef CAS.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed. Engl., 2007, 46, 7636–7639 CrossRef CAS PubMed.
- N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang and J. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef CAS PubMed.
- A. Corma Canos, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef PubMed.
- K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS.
- W. Schutyser, S. Van Den Bosch, T. Renders, T. De Boe, S. F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Green Chem., 2015, 17, 5035–5045 RSC.
- S. F. Koelewijn, S. Van Den Bosch, T. Renders, W. Schutyser, B. Lagrain, M. Smet, J. Thomas, W. Dehaen, P. Van Puyvelde, H. Witters and B. F. Sels, Green Chem., 2017, 19, 2561–2570 RSC.
- M. S. Kathalewar, P. B. Joshi, A. S. Sabnis and V. C. Malshe, RSC Adv., 2013, 3, 4110–4129 RSC.
- F. D. Bobbink, W. Gruszka, M. Hulla, S. Das and P. J. Dyson, Chem. Commun., 2016, 52, 10787–10790 RSC.
- M. Honda, M. Tamura, K. Nakao, K. Suzuki, Y. Nakagawa and K. Tomishige, ACS Catal., 2014, 4, 1893–1896 CrossRef CAS.
- L. Maisonneuve, A. S. More, S. Foltran, C. Alfos, F. Robert, Y. Landais, T. Tassaing, E. Grau and H. Cramail, RSC Adv., 2014, 4, 25795–25803 RSC.
- M. Bähr and R. Mülhaupt, Green Chem., 2012, 14, 483–489 RSC.
- R. Pathak, M. Kathalewar, K. Wazarkar and A. Sabnis, Prog. Org. Coat., 2015, 89, 160–169 CrossRef CAS.
- K. Kaneda, S. Haruna, T. Imanaka, M. Hamamoto, Y. Nishiyama and Y. Ishii, Tetrahedron Lett., 1992, 33, 6827–6830 CrossRef CAS.
- J. A. Galbis, M. D. G. García-Martín, M. V. De Paz and E. Galbis, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS PubMed.
- X. Feng, A. J. East, W. B. Hammond, Y. Zhang and M. Jaffe, Polym. Adv. Technol., 2011, 22, 139–150 CrossRef CAS.
- J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- J. Martínez, J. Fernández-Baeza, L. F. Sánchez-Barba, J. A. Castro-Osma, A. Lara-Sánchez and A. Otero, ChemSusChem, 2017, 10, 2886–2890 CrossRef PubMed.
- X. Shen, X. Liu, J. Wang, J. Dai and J. Zhu, Ind. Eng. Chem. Res., 2017, 56, 8508–8516 CrossRef CAS.
- F. D. Bobbink, J. Zhang, Y. Pierson, X. Chen and N. Yan, Green Chem., 2015, 17, 1024–1031 RSC.
- M. Pelckmans, T. Renders, S. Van de Vyver and B. F. Sels, Green Chem., 2017, 19, 5303–5331 RSC.
- M. Pelckmans, W. Vermandel, F. Van Waes, K. Moonen and B. F. Sels, Angew. Chem., Int. Ed., 2017, 56, 14540–14544 CrossRef CAS PubMed.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- Y. Pierson, X. Chen, F. D. Bobbink, J. Zhang and N. Yan, ACS Sustainable Chem. Eng., 2014, 2, 2081–2089 CrossRef CAS.
- V. Froidevaux, C. Negrell, S. Caillol, J. P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- R. Besse, S. Auvergne, G. Carlotti, B. Boutevin, S. Otazaghine, J. Caillol and B. Pascault, React. Funct. Polym., 2013, 73, 588–594 CrossRef.
- A. Cornille, G. Michaud, F. Simon, S. Fouquay, R. Auvergne, B. Boutevin and S. Caillol, Eur. Polym. J., 2016, 84, 404–420 CrossRef CAS.
- M. Blain, L. Jean-Gérard, R. Auvergne, D. Benazet, S. Caillol and B. Andrioletti, Green Chem., 2014, 16, 4286–4291 RSC.
- W. Schutyser, T. Renders, S. Van Den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- N. Kihara and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2765–2773 CrossRef CAS.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- G. Beniah, W. H. Heath and J. M. Torkelson, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3347–3351 CrossRef CAS.
- T. J. Farmer, J. W. Comerford, A. Pellis and T. Robert, Polym. Int., 2018, 67, 775–789 CrossRef CAS.
- Q. W. Lu, T. R. Hoye and C. W. Macosko, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2310–2328 CrossRef CAS.
- M. Cregut, M. Bedas, M. J. Durand and G. Thouand, Biotechnol. Adv., 2013, 31, 1634–3647 CrossRef CAS PubMed.
- A. Cornille, S. Dworakowska, D. Bogdal, B. Boutevin and S. Caillol, Eur. Polym. J., 2015, 66, 129–138 CrossRef CAS.
- A. Cornille, R. Auvergne, O. Figovsky, B. Boutevin and S. Caillol, Eur. Polym. J., 2017, 87, 535–552 CrossRef CAS.
- C. Duval, N. Kébir, R. Jauseau and F. Burel, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 758–764 CrossRef CAS.
- M. Blain, H. Yau, L. Jean-Gérard, R. Auvergne, D. Benazet, P. R. Schreiner, S. Caillol and B. Andrioletti, ChemSusChem, 2016, 9, 2269–2272 CrossRef CAS PubMed.
- V. M. Lombardo, E. A. Dhulst, E. K. Leitsch, N. Wilmot, W. H. Heath, A. P. Gies, M. D. Miller, J. M. Torkelson and K. A. Scheidt, Eur. J. Org. Chem., 2015, 2791–2795 CrossRef CAS.
- A. Bossion, R. H. Aguirresarobe, L. Irusta, D. Taton, H. Cramail, E. Grau, D. Mecerreyes, C. Su, G. Liu, A. J. Müller and H. Sardon, Macromolecules, 2018, 51, 5556–5566 CrossRef CAS.
- S. Sopeña, V. Laserna, W. Guo, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Adv. Synth. Catal., 2016, 358, 2172–2178 CrossRef.
- S. Gennen, B. Grignard, T. Tassaing, C. Jérôme and C. Detrembleur, Angew. Chem., Int. Ed., 2017, 56, 10394–10398 CrossRef CAS PubMed.
- L. Han, J. Dai, L. Zhang, S. Ma, J. Deng, R. Zhang and J. Zhu, RSC Adv., 2014, 4, 49471–49477 RSC.
- Y. Liu, W. M. Ren, K. K. He, W. Z. Zhang, W. B. Li, M. Wang and X. B. Lu, J. Org. Chem., 2016, 81, 8959–8966 CrossRef CAS PubMed.
- W. Guo, V. Laserna, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Chem. – Eur. J., 2016, 22, 1722–1727 CrossRef CAS PubMed.
- B. D. Ulery, L. S. Nair and C. T. Laurencin, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 832–864 CrossRef CAS PubMed.
- Q. Liu, L. Jiang, R. Shi and L. Zhang, Prog. Polym. Sci., 2012, 37, 715–765 CrossRef CAS.
- Y. Hong, J. Guan, K. L. Fujimoto, R. Hashizume, A. L. Pelinescu and W. R. Wagner, Biomaterials, 2010, 31, 4249–4258 CrossRef CAS PubMed.
- A. A. Shah, F. Hasan, A. Hameed and S. Ahmed, Biotechnol. Adv., 2008, 26, 246–265 CrossRef CAS PubMed.
- A. Zgoła-Grześkowiak, T. Grześkowiak, J. Zembrzuska and Z. Łukaszewski, Chemosphere, 2006, 64, 803–809 CrossRef PubMed.
- G. T. Howard, Int. Biodeterior. Biodegrad., 2002, 49, 245–252 CrossRef CAS.
- N. Lucas, C. Bienaime, C. Belloy, M. Queneudec, F. Silvestre and J. E. Nava-Saucedo, Chemosphere, 2008, 73, 429–442 CrossRef CAS PubMed.
- B. Liu, Biomacromolecules, 2018, 19, 1783–1803 CrossRef PubMed.
- T. Garrison, A. Murawski and R. Quirino, Polymers, 2016, 8, 262 CrossRef.
- A. Cornille, C. Guillet, S. Benyahya, C. Negrell, B. Boutevin and S. Caillol, Eur. Polym. J., 2016, 84, 873–888 CrossRef CAS.
- D. Carta, G. Cao and C. D’Angeli, Environ. Sci. Pollut. Res., 2003, 10, 390–394 CrossRef CAS PubMed.
- M. Genta, F. Yano, Y. Kondo, W. Matsubara and S. Oomoto, Mitsubishi Heavy Ind. Ltd., Technol. Rev., 2003, 40, 1–4 Search PubMed.
- F. R. Veregue, C. T. Pereira da Silva, M. P. Moisés, J. G. Meneguin, M. R. Guilherme, P. A. Arroyo, S. L. Favaro, E. Radovanovic, E. M. Girotto and A. W. Rinaldi, ACS Sustainable Chem. Eng., 2018, 6, 12017–12024 CrossRef CAS.
- S. Westhues, J. Idel and J. Klankermayer, Sci. Adv., 2018, 4, eaat9669 CrossRef PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- P. Izák, F. D. Bobbink, M. Hulla, M. Klepic, K. Friess, Š. Hovorka and P. J. Dyson, ChemPlusChem, 2018, 83, 7–18 CrossRef.
- A. Barthel, Y. Saih, M. Gimenez, J. D. A. Pelletier, F. E. Kühn, V. D’Elia and J.-M. Basset, Green Chem., 2016, 18, 3116–3123 RSC.
- J. Song, B. Zhang, P. Zhang, J. Ma, J. Liu, H. Fan, T. Jiang and B. Han, Catal. Today, 2012, 183, 130–135 CrossRef CAS.
- Y. Hu, J. Song, C. Xie, H. Wu, Z. Wang, T. Jiang, L. Wu, Y. Wang and B. Han, ACS Sustainable Chem. Eng., 2018, 6, 11228–11234 CrossRef CAS.
- J. Tharun, Y. Hwang, R. Roshan, S. Ahn, A. C. Kathalikkattil and D.-W. Park, Catal. Sci. Technol., 2012, 2, 1674–1680 RSC.
- S. Das, F. D. Bobbink, S. Bulut, M. Soudani and P. J. Dyson, Chem. Commun., 2016, 52, 2497–2500 RSC.
- A. Laca, A. Laca and M. Díaz, J. Environ. Manage., 2017, 197, 351–359 CrossRef CAS PubMed.
- N. von der Assen, J. Jung and A. Bardow, Energy Environ. Sci., 2013, 6, 2721–2734 RSC.
- J. Artz, T. E. Müller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |