
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolar-driven CO2 to CO reduction utilizing H2O as an electron donor by earth-abundant Mn–bipyridine complex and Ni-modified Fe-oxyhydroxide catalysts activated in a single-compartment reactor†
Takeo
Arai
 *,
Shunsuke
Sato
,
Keita
Sekizawa
,
Tomiko M.
Suzuki
and
Takeshi
Morikawa
*,
Shunsuke
Sato
,
Keita
Sekizawa
,
Tomiko M.
Suzuki
and
Takeshi
Morikawa
Toyota Central Research and Development Laboratories, Inc., 41-1 Yokomichi, Nagakute, Aichi 480-1192, Japan. E-mail: takeo–arai@mosk.tytlabs.co.jp
First published on 3rd December 2018
Abstract
Photoelectrochemical CO2 to CO reduction was demonstrated with 3.4% solar-to-chemical conversion efficiency using polycrystalline silicon photovoltaic cells connected with earth-abundant catalysts: a manganese complex polymer for CO2 reduction and iron oxyhydroxide modified with a nickel compound for water oxidation. The system operated around neutral pH in a single-compartment reactor.
The development of artificial photosynthesis systems that can reduce carbon dioxide (CO2) to useful and sustainable organic chemicals using sunlight is important to mitigate worldwide climate change and fossil fuel shortages. A photoelectrochemical approach is one candidate method to reduce CO2 through the use of solar energy. Many reports on photoelectrochemical CO2 reduction system have been reported.1–7 We also have reported a monolithic artificial photosynthesis device with molecular and metal-oxide catalysts attached with a semiconductor photosensitizer, which can produce formate with a solar to chemical conversion efficiency of 4.6% in a simple single-compartment reactor.4,5 However, the device requires noble metal catalysts, such as a ruthenium complex polymer as a CO2 reduction catalyst, and iridium oxide as a water oxidation catalyst. In view of future practical applications, such a system requires efficient catalysts that consist of inexpensive and abundant elements. There have been several reports on systems composed of earth-abundant metal catalysts.6,7 Carbon monoxide (CO) is a raw material of high industrial value. Solar-driven CO2 to CO conversion using a SnO2 modified CuO nanowire electrode for both CO2 reduction and the oxygen evolution reaction (OER) has been reported by Schreier et al.6 14.4% of the total solar-to-fuel efficiency was observed by the combination of GaInP/GaInAs/Ge solar cells as a light absorber. Urbain et al. observed a solar-to-syngas conversion efficiency of 4.3% using a system consisting of a copper-zinc cathode for CO2 reduction to CO and a photoanode comprised of nickel foam catalyst for H2O oxidation and a heterojunction with intrinsic thin layer (HIT) solar cell (silicon solar cell) as a light absorber.7 A two-compartment reactor separated by a bipolar membrane was used in these systems. The theoretical potential difference of a perfectly permselective bipolar membrane for the generation of H+ and OH− was reported to be 0.83 V by calculation,8 and this will be an additional required potential to operate the CO2 electrolyzer. These systems used two types of electrolyte to reduce the operating potential by the chemical bias accompanied with the pH difference between catholyte and anolyte. However, the pH differences reported in these systems were around 6, which suggests a potential loss of 0.47 V by the membrane. On the other hand, we have reported a simple CO2 reduction system in a single-compartment reactor without an expensive membrane, which has the advantage of being a simplified system.4 Herein, we demonstrate an example of an earth-abundant CO2 electrolyzer in a single-compartment reactor by the combination of a manganese complex catalyst and iron oxyhydroxide. The Mn-complex is a candidate CO2 reduction catalyst that consists of earth-abundant elements. Many Mn-complexes have been reported for electrochemical or photocatalytic CO2 reduction.9–12 We have reported that a Mn-complex polymer (hereinafter referred to as [Mn–MeCN]) on multi-walled carbon nanotubes (MWCNTs) facilitated electrocatalytic CO2 reduction in a mixed aqueous solution of potassium borate and sulfate (hereinafter referred to as KBB) with near neutral pH, while the bare Mn-complex did not function as a catalyst in the same solution.11 The overpotential of electrocatalytic CO2 reduction over [Mn–MeCN] was significantly reduced to 100 mV by the synergetic effect of the MWCNT support and K+ cations. Iron oxyhydroxide (FeOOH) has been reported as an earth-abundant OER catalyst.13–19 We have recently reported akaganeite-type iron oxyhydroxide hyperfine nanorod doped with 1 at% nickel (β-FeOOH:Ni) surface-modified with 21 at% amorphous nickel hydride (a-Ni(OH)2) in which Ni amounts are defined as Ni/Fe ratio (hereinafter referred to as β-FeOOH:Ni/a-Ni(OH)2).19 β-FeOOH:Ni/a-Ni(OH)2 exhibited higher performance than other Fe-based oxide and oxyhydroxide materials, and an overpotential of 430 mV (1.66 V vs. RHE) at 10 mA cm−2 was observed during the OER under strong alkaline conditions. However, Fe-based materials generally have low activity at near neutral pH. Therefore, the utilization of β-FeOOH:Ni/a-Ni(OH)2 as an OER catalyst in near neutral pH is a challenging and important approach. To conduct CO2 reduction in a single-compartment reactor, the β-FeOOH:Ni/a-Ni(OH)2 catalyst was used in KBB aqueous solution and the pre-treatment of β-FeOOH:Ni/a-Ni(OH)2 in strong alkaline condition was found to be effective to maintain the OER activity in near neutral solution. Photoelectrochemical CO2 reduction was subsequently demonstrated by combining the earth-abundant CO2 electrolyzer with photovoltaic cells.
The current–potential characteristics of β-FeOOH:Ni/a-Ni(OH)2 (see ESI† for detail) on carbon paper were evaluated in 1 M KOH (pH 13.6), 0.1 M KOH (pH 12.9) and 0.1 M KBB (pH 6.9) aqueous solutions (Fig. 1(a)). The apparent surface area (ASA) of the β-FeOOH:Ni/a-Ni(OH)2 electrode was 2 cm2. The overpotential during the OER increased significantly with a decrease in the pH of the electrolyte from alkaline to neutral. The same pH dependence has been reported for the activity of a Fe-free NiOOH catalyst.20 Diaz-Morales et al. identified the active species as a deprotonated γ-NiOOH surface phase that was generated by the electrochemical oxidation of Ni(OH)2. They have proposed a mechanism for the OER on NiOOH that involves deprotonation of the polymeric hydrous nickel oxyhydroxide by hydroxide ion to form a superoxo-type intermediate (NiOO−) that acts as preferential oxygen precursor at pH > 11. NiOOH was described as an unsuitable electrocatalyst for applications in neutral or moderately alkaline pH (in the range 7–11). In the case of β-FeOOH:Ni/a-Ni(OH)2,19 it is considered that the amorphous Ni(OH)2 on β-FeOOH:Ni influences the pH dependence of the OER activity. Cathodic peaks were observed at +1.36 V and +1.40 V (vs. RHE) in 1 M and 0.1 M KOH solutions, respectively (Fig. 1(b)). The β-FeOOH:Ni/a-Ni(OH)2 electrode after use in 1 M KOH solution also showed a cathodic peak at +1.49 V (vs. RHE) in 0.1 M KBB solution, while a broad and ambiguous peak was observed for the bare β-FeOOH:Ni/a-Ni(OH)2 electrode in 0.1 M KBB solution. Fig. 1(c) shows the results of current–potential measurements repeated in 0.1 M KBB solution for the bare β-FeOOH:Ni/a-Ni(OH)2 electrode. The anodic current for the OER was decreased by repeating the measurement. The overpotential was increased during the three times potential sweep. The β-FeOOH:Ni/a-Ni(OH)2 electrode after used in the 1 M KOH solution showed stable current–potential characteristics, even in 0.1 M KBB solution (Fig. 1(d)). The pH dependence of the Ni–Fe oxyhydroxide catalyst supported on Vulcan XC-72r (NiFeOx/C) has been investigated by Görlin et al.16 They reported that a 0.1 M KOH (pH 13) pre-cycled NiFeOx/C electrode showed a rapid loss of catalytic OER activity in 0.1 M phosphate buffer (pH 7) solution during cyclic voltammetry measurements. Ni(OH)2/NiOOH redox peaks were observed around +1.4 V and +1.5 V (vs. RHE) at pH 13 and pH 7, respectively. The potentials of cathodic peaks shown in Fig. 1(b) were almost correspond to the results of Görlin et al. at each pH, which suggests these are Ni(OH)2/NiOOH redox peaks. Ni(OH)2/NiOOH (or NiII/NiIII) redox transition is considered to be strongly related to the OER activity in several reports.16,17,20 According to the Pourbaix diagrams reported by Beverskog et al., the formation of Ni(OH)2 seems difficult around neutral pH.21 Therefore, it is inferred that the pre-formation of the Ni(OH)2/NiOOH redox couple using β-FeOOH:Ni/a-Ni(OH)2 under strong alkaline conditions is important to maintain the OER activity under neutral conditions. According to Görlin et al.,16 the current–potential characteristics of the 0.1 M KOH pre-cycled NiFeOx/C electrode reveals a Ni(OH)2/NiOOH redox peak in 0.1 M phosphate buffer. However, the OER activity was gradually decreased with repeated cyclic voltammetry measurements and the Ni(OH)2/NiOOH redox peak diminished due to a loss of Ni atoms. Lee et al. reported that PO43− tends to destroy the structure of Ni(OH)2.22 It has also been reported that a 0.1 M KOH pre-cycled NiFeOx/C electrode showed stable OER activity in 0.1 M borate buffer (pH 9.2).16 According to these reports, the important factor to maintain OER activity under neutral conditions is not only the formation of the Ni(OH)2/NiOOH redox couple, but also the presence of borate and the absence of phosphate.
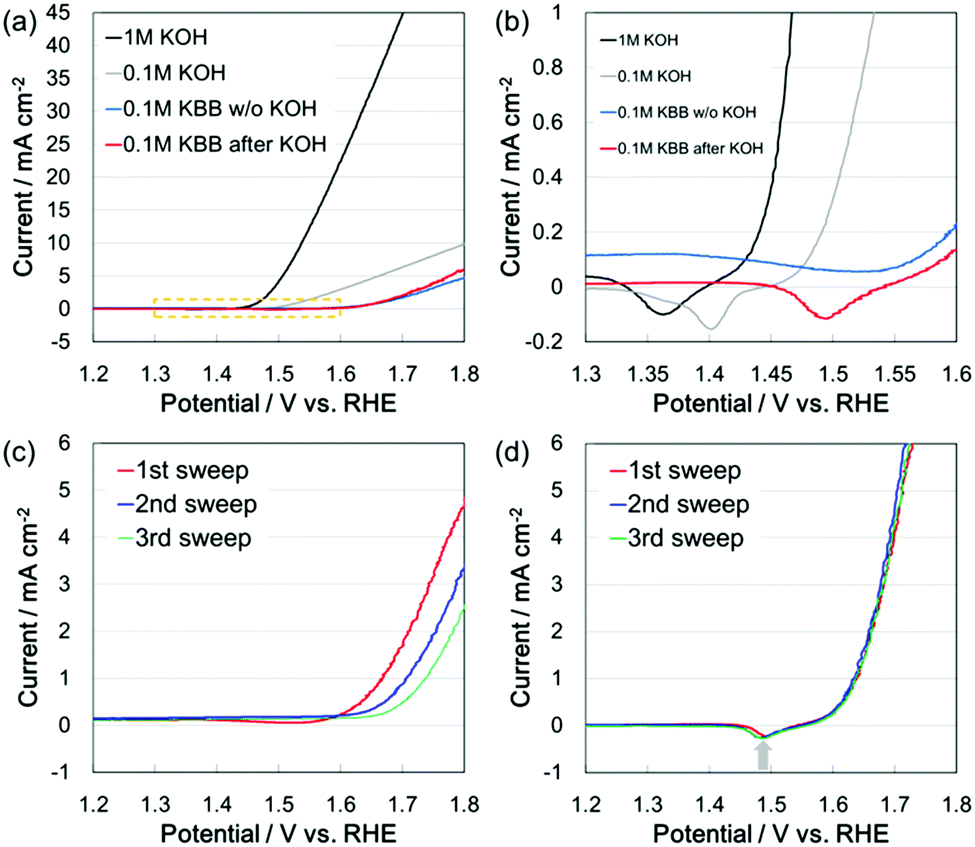 | ||
| Fig. 1 Current–potential characteristics of the β-FeOOH:Ni/a-Ni(OH)2 electrode in various electrolytes. (a) Comparison of the current–potential characteristics in 1 M KOH (pH 13.6), 0.1 M KOH (pH 12.9) and 0.1 M KBB (pH 6.9) solutions. (b) Enlargement of the yellow dotted line area in (a). (c) Current–potential characteristics of the bare β-FeOOH:Ni/a-Ni(OH)2 electrode in 0.1 M KBB solution. (d) Current–potential characteristics of the β-FeOOH:Ni/a-Ni(OH)2 electrode in 0.1 M KBB after use in 1 M KOH solution. | ||
The OER activity of the β-FeOOH:Ni/a-Ni(OH)2 electrode was slightly improved by modification with single-walled carbon nanotubes on carbon paper (see the results for Fig. S5, ESI†). The 0.1 M KOH pre-cycled β-FeOOH:Ni/a-Ni(OH)2 electrode (ASA: 2 cm2) was combined with a [Mn–MeCN] electrode (ASA: 3.24 cm2, see ESI† for detail) to compose a single-compartment CO2 electrolyzer (Fig. S3, ESI†). A series of six polycrystalline silicon photovoltaic cells (hereinafter referred to as poly-Si cells) were selected as a light absorber because it was cheap and commercially available. The current–potential characteristics of the CO2 electrolyzer and poly-Si cells are shown in Fig. 2(a). The cross-point that indicates the operation point of the CO2 electrolyzer and poly-Si cells was estimated to be 2.19 V and 5.1 mA. The CO2 electrolyzer was connected with poly-Si cells to convert light energy to chemical energy. The current–time measurement was conducted under solar simulated light irradiation. The applied potentials of the cathode and anode by poly-Si cells were −1.03 V and +1.12 V (vs. Ag/AgCl), respectively. The observed current was ca. 5 mA, which corresponds to the operation current estimated from the current–potential characteristics, and it was stable during 4 hours of measurement (Fig. 2(b)). Time courses for the amount of gaseous products during CO2 reduction are shown in Fig. 2(c). The main products were CO and H2 for the reduction reaction and O2 for the oxidation reaction. The amount of other CO2 reduction products was negligible, and the ratio of reduction products CO and H2 were 82% and 18%, respectively (see ESI† for a margin error). The amount of O2 was approximately stoichiometric with the CO and H2 production. It is worth noting that CO production selectivity of over 80% was observed while oxygen was generated from water in a single-compartment reactor. CO2 reduction is generally disturbed by the reduction of O2 as a competitive reaction. Carbon materials are reported to act as catalyst supports that prevent O2 reduction4 and promote CO2 reduction in a combination with metal complex catalysts.11,23 The carbon source for the generation of CO over [Mn–MeCN] was identified as CO2 by isotope tracer analysis in our previous study.11 Therefore, the present CO was generated by the reduction of CO2 using H2O as an electron donor. The turnover number for CO production was estimated to be 120 during 4 hours reaction. The solar to chemical energy conversion efficiency for CO production was calculated to be 3.4% (see ESI† for details) and the efficiency for CO and H2 production was 4.1%. The efficiency was slightly low compared with the solar-to-syngas efficiency 4.3% when using a silicon HIT cell.6,7 However, this value is strongly dependent on the solar-to-electricity conversion efficiency of the photovoltaic cell; the efficiency of the poly-Si cell was 7.1%, while that of typical HIT solar cells is 16–19%. Therefore, 4.1% efficiency achieved in the single-compartment reactor without membrane by the combination of these earth-abundant Mn and Fe catalysts and the inexpensive poly-Si cells is technically advantageous.
 | ||
| Fig. 2 Results of solar driven CO2 reduction using a CO2 electrolyzer and poly-Si cells. The CO2 electrolyzer consisted of a [Mn–MeCN] electrode (ca. 3.24 cm2) for CO2 reduction and a β-FeOOH:Ni/a-Ni(OH)2 electrode (ca. 2 cm2) for H2O oxidation in a single-compartment reactor. The electrolyte was CO2 saturated 0.1 M KBB solution. Poly-Si cells are the series of 6 polycrystalline silicon photovoltaic cells. (a) Current–potential characteristics of the CO2 electrolyzer and poly-Si cells under simulated solar light (1 sun, AM1.5, 1.6 cm2). Time courses of (b) the current under potentiostatic conditions (applied bias 0 V) and (c) amount of gaseous products observed during solar driven CO2 reduction using the combination of CO2 electrolyzer and poly-Si cells. | ||
Solar-driven CO2 reduction was also conducted with the combination of [Mn–MeCN], Ni(OH)2-deposited β-FeOOH:Ni/a-Ni(OH)2 and a triple-junction amorphous silicon photovoltaic cell (3jn-a-Si cell) as a light absorber (Fig. S4, ESI†). Tandem photovoltaic cells such as 3jn-a-Si provide high voltages (Fig. S6, ESI†) that exceed 1.33 V of the theoretical potential to reduce CO2 to CO using water as an electron donor. The tandem photovoltaic light absorber is advantageous because CO2 can be reduced without series connection and it is suitable for future fabrication of a monolithic artificial photosynthesis system.4 Conventional Si cells generally have an open circuit potential of 0.7 V and a series of over 3 photovoltaic cells are thus required to conduct CO2 reduction to CO including the overpotential of the catalysts. Series connection of photovoltaic cells generally leads to a loss of voltage and current due to the resistance caused by the non-uniformity of solar irradiation to each cell, and a conditioner is thus required to control the voltage. The solar-to-chemical energy conversion efficiency using the 3jn-a-Si cell as a light absorber was estimated to be 1.0% for CO generation. The current efficiencies for CO, H2 and O2 were 84.7%, 18.4% and 97.8%, respectively (Fig. S7, ESI†). The product ratios were almost the same as those observed for the poly-Si cells. Photoelectrochemical CO2 reduction to CO using the same light absorber has been reported by Sugano et al.,24 where the system was composed of an Au nanoparticle cathode and a photoanode that combined a 3jn-a-Si cell and a CoOx catalyst. A two-compartment reactor separated by an anion exchange membrane was used for the wired photovoltaic-photoelectrochemical cell system. The solar-to-CO conversion efficiency was less than 1.3% when both the cathode and anode were bubbled with CO2 during irradiation. Comparable efficiency was achieved using the earth-abundant Mn-complex and β-FeOOH:Ni catalysts instead of Au and CoOx. This result suggests a future possibility to realize an ultimately simplified monolithic artificial photosynthesis device to produce only gaseous products with earth-abundant catalysts and a photoabsorber.
In this study, we demonstrated a CO2 electrolyzer utilizing earth-abundant catalysts of a Mn metal complex and nickel-modified akaganeite-type iron oxyhydroxide in a single-compartment reactor. Photoelectrochemical CO2 reduction to CO was conducted by combination with photovoltaic cells. This is also the first example of photoelectrochemical CO2 reduction utilizing an earth-abundant metal complex catalyst. Over 80% current efficiency for CO production was achieved with a solar-to-chemical conversion efficiency of 3.4%, while oxygen was generated as an oxidation product in the same compartment. A 3jn-a-Si cell was also available as a light absorber to reduce CO2, which suggests the possibility to realize a monolithic artificial photosynthesis composed of earth-abundant catalysts. It is supposed that modification of the Mn complex on MWCNTs contributes to a high reaction selectivity and low operation potential, as previously reported.11β-FeOOH:Ni was stabilized even under near-neutral pH conditions by pre-treatment in strong alkaline solution, which suggests that the formation of the Ni(OH)2/NiOOH redox couple is important for the OER at neutral pH.
This work was supported in part by the Advanced Catalytic Transformation Program for Carbon Utilization (ACT-C, Grant Number JPMJCR12ZA) of the Japan Science and Technology Agency (JST). The authors would like to thank H. Uchiyama for experimental support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- T. Haining, ChemSusChem, 2015, 8, 3746–3759 CrossRef PubMed.
- N. Zhang, R. Long, C. Gao and Y. J. Xiong, Sci. China Mater., 2018, 61, 771–805 CrossRef.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 RSC.
- S. Sato, T. Arai and T. Morikawa, Nanotechnology, 2018, 29, 034001 CrossRef PubMed.
- M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2, 17087 CrossRef CAS.
- F. Urbain, P. Tang, N. M. Carretero, T. Andreu, L. G. Gerling, C. Voz, J. Arbiol and J. R. Morante, Energy Environ. Sci., 2017, 10, 2256–2266 RSC.
- K. Nagasubramanian, F. P. Chlanda and K.-J. Liu, J. Membr. Sci., 1977, 2, 109–124 CrossRef CAS.
- M. Stanbury, J.-D. Compain and S. Chardon-Noblat, Coord. Chem. Rev., 2018, 361, 120–137 CrossRef CAS.
- A. Sinopoli, N. T. La Porte, J. F. Martinez, M. R. Wasielewski and M. Sohail, Coord. Chem. Rev., 2018, 365, 60–74 CrossRef CAS.
- S. Sato, K. Saita, K. Sekizawa, S. Maeda and T. Morikawa, ACS Catal., 2018, 8, 4452–4458 CrossRef CAS.
- D. C. Grills, M. Z. Ertem, M. McKinnon, K. T. Ngo and J. Rochford, Coord. Chem. Rev., 2018, 374, 173–217 CrossRef CAS.
- J. A. Seabold and K.-S. Choi, J. Am. Chem. Soc., 2012, 134, 2186–2192 CrossRef CAS PubMed.
- W. D. Chemelewski, H.-C. Lee, J.-F. Lin, A. J. Bard and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 2843–2850 CrossRef CAS PubMed.
- O. Diaz-Morales, I. Ledezma-Yanez, M. T. M. Koper and F. Calle-Vallejo, ACS Catal., 2015, 5, 5380–5387 CrossRef CAS.
- M. Görlin, M. Gliech, J. F. de Araújo, S. Dresp, A. Bergmann and P. Strasser, Catal. Today, 2016, 262, 65–73 CrossRef.
- M. Görlin, J. Ferreira de Araújo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef PubMed.
- T. M. Suzuki, T. Nonaka, A. Suda, N. Suzuki, Y. Matsuoka, T. Arai, S. Sato and T. Morikawa, Sustainable Energy Fuels, 2017, 1, 636–643 RSC.
- T. M. Suzuki, T. Nonaka, K. Kitazumi, N. Takahashi, S. Kosaka, Y. Matsuoka, K. Sekizawa, A. Suda and T. Morikawa, Bull. Chem. Soc. Jpn., 2018, 91, 778–786 CrossRef CAS.
- O. Diaz-Morales, D. Ferrus-Suspedra and M. T. M. Koper, Chem. Sci., 2016, 7, 2639–2645 RSC.
- B. Beverskog and I. Puigdomenech, Corros. Sci., 1997, 39, 969–980 CrossRef CAS.
- M. Lee, H. S. Jeon, S. Y. Lee, H. Kim, S. J. Sim, Y. J. Hwang and B. K. Min, J. Mater. Chem. A, 2017, 5, 19210–19219 RSC.
- S. Sato, T. Arai and T. Morikawa, J. Photochem. Photobiol., A, 2016, 327, 1–5 CrossRef CAS.
- Y. Sugano, A. Ono, R. Kitagawa, J. Tamura, M. Yamagiwa, Y. Kudo, E. Tsutsumi and S. Mikoshiba, RSC Adv., 2015, 5, 54246–54252 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc07900e |
| This journal is © The Royal Society of Chemistry 2019 |