
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent-free lithium and sodium containing electrolytes based on pseudo-delocalized anions†
Juan
Forero-Saboya‡
 a,
Elham
Hosseini-Bab-Anari‡
b,
Muhammad E.
Abdelhamid
a,
Kasper
Moth-Poulsen
b and
Patrik
Johansson
*ac
a,
Elham
Hosseini-Bab-Anari‡
b,
Muhammad E.
Abdelhamid
a,
Kasper
Moth-Poulsen
b and
Patrik
Johansson
*ac
aDepartment of Physics, Chalmers University of Technology, SE-412 96 Gothenburg, Sweden. E-mail: patrik.johansson@chalmers.se
bDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, SE-412 96 Gothenburg, Sweden
cALISTORE – European Research Institute, CNRS FR 3104, Hub de l’Energie, Rue Baudelocque, 80039 Amiens Cedex, France
First published on 10th December 2018
Abstract
Mixing the standard battery salt LiTFSI with various Li-salts of novel pseudo-delocalized organic anions [N(CH3)2((CH2)nSO3)((CH2)mSO3)]− (MMnm11), results in super-cooled solvent-free liquid electrolytes with glass transition temperatures of ca. 50 °C. Synthesis routes and full chemical characterisation of the new pseudo-delocalized anions are presented, as well as phase and thermal stabilities. The ion conductivities and electrochemical stabilities are evaluated towards lithium and sodium battery application.
Conventional lithium-ion battery (LIB) electrolytes are composed of lithium salts with weakly coordinating anions, such as hexafluorophosphate1 (PF6−), tetrafluoroborate2 (BF4−), bis(oxalate)borate3 (BOB), or bis(trifluoromethanesulfonyl)imide4 (TFSI), dissolved in a mixture of organic solvents (often linear and cyclic carbonates) at ca. 1 M concentrations.5,6 Their wide electrochemical stability windows (ESWs) allow the use of high voltage positive electrodes, such as LiCoO2 (4 V vs. Li+/Li), and graphite negative electrodes (0.1 V vs. Li+/Li), by virtue of the creation of a solid electrolyte interphase (SEI). This, together with the large capacities of the electrodes, enable a very high specific energy. But the safety of LIBs remains a major concern mainly due to the highly flammable organic solvents used.
The combination of ever higher energy densities and voltages of the cells with the highly flammable organic solvents7 directs particular attention towards creating novel electrolytes with improved safety characteristics. For example, replacing the organic solvents by an ionic liquid matrix improves the thermal stability drastically and decreases the flammability,8 but at the same time, both the lithium transference number and the electrolyte fluidity often decreases, thus reducing the electrochemical performance.
All solid-state batteries (ASSBs),9,10 in which the conventional electrolytes have been replaced by a solid-state electrolyte (SSE), also claim to provide remarkable safety characteristics. However, most SSEs suffer from low room temperature ionic conductivities, often three orders of magnitude lower than liquid electrolytes, hence they usually operate at elevated temperatures (ca. 80 °C),11 even if there exist polymer electrolytes with acceptable ionic conductivities at room temperature (1 mS cm−1). As a result, however, the concept of high-temperature lithium-ion batteries (HT-LIB) has emerged.12
Another alternative is “thermal” batteries, operating between 350 °C and 550 °C and employing molten salt mixtures as liquid solvent-free electrolytes, with high thermal stability, low flammability, and high ionic conductivity.11 In order to utilize this concept for LIBs, the melting temperature must decrease significantly – but even if these electrolytes do not reach the conductivities needed at room temperature, they might be applied in HT-LIBs. This has been achieved by combining two or three alkali cation salts with a common weakly coordinating anion, such as TFSI,13,14 bis(fluorosulfonyl)imide (FSI),15 or triflate.16 Such eutectic mixtures have promising ionic conductivities and form stable SEIs on typical LIB electrode materials. The molar fraction of Li+ is, however, quite low, which severely limits the rate capability of the cells despite the elevated operating temperature.17 Additionally, the presence of other alkali metal cations may interfere with the electrochemistry by unwanted plating or co-intercalation. The approach has its origin in enhanced ion conductivities by the “mixed alkali effect” of glassy and polymeric electrolytes, explored for batteries in the early 1990's.18,19
Herein, an alternative strategy is explored to create solvent-free electrolytes; mono-cationic–di-anionic mixtures, i.e. mixing two Li-salts with different anions, using the unique design of three different pseudo-delocalized (i.e. the negative charge delocalized by being localized at several central in the structure) anions [N(CH3)2((CH2)nSO3)((CH2)mSO3)]− (MMnm11, Fig. 1)20,21 – aiming primarily at HT-LIB application. We explore the previously reported lithium and sodium salts of MM4411 together with a new symmetric anion (MM3311) as well as an asymmetric anion (MM3411), all synthesized with high purity and excellent yield in three consecutive steps from readily available pre-cursors with minimal usage of solvent on multiple gram scale, expected to be scaled-up to kg scale without any practical difficulties. An electrolyte based 100% on salts is likely to initially be more expensive than a 1 M Li-salt liquid electrolyte, but improved battery safety and life-length may result. The here presented anions are likely to be cheap to produce based on the pre-cursors. The synthesis details and the full characterisation is found in the ESI.†
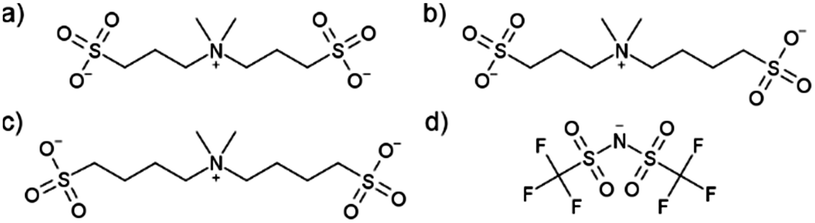 | ||
| Fig. 1 Chemical structures of the anions employed: (a) MM3311: 3,3′-(dimethylammonio)bis(propane-1-sulfonate), (b) MM3411: 4-(dimethyl(3-sulfonatopropyl)ammonio)butane-1-sulfonate, (c) MM4411: 4,4′-(dimethylammonio)bis(butane-1-sulfonate), and (d) TFSI: bis(trifluoromethylsulfonyl)imide. | ||
First, the thermal stabilities of the MMnm11 salts were assessed, whereafter their mixtures with TFSI salts were explored by constructing a series of quasi-phase diagrams highlighting the glass transition temperatures (Tgs) and melting points. Second, the ion conductivities as a function of both temperature and composition were studied and analysed using free volume theory. Finally, the lithium-only composition with the lowest Tg was selected for proof-of-concept electrochemical stability tests aiming towards lithium battery application.
The pure salts LiMM3311 and LiMM3411 exhibit very similar thermal behaviour. The first weight loss of ∼1.8% at 140 °C is most likely due to residual water from the synthesis (Fig. 2a) as confirmed by TGA-FTIR evolved gas analysis (Fig. 2b), where the mass loss clearly observed in the derivate of the thermogravimetric signal (dTG) corresponds to an increased H2O bend band signal. Both salts decompose at ca. 280 °C, as observed in the dTG signal and an increased C–H stretching band intensity.
 | ||
| Fig. 2 (a) TGA traces of the LiMMnm11 salts (the corresponding NaMMnm11 salts present similar behaviour (Fig. S11, ESI†)), (b) FTIR band intensities evolution with temperature for the thermal analysis of LiMM3411, and (c) first derivative of the TGA signal of LiMM3411. | ||
In stark contrast the LiMM4411 salt decomposes much later, at ca. 350 °C, implying higher stability of butylsulfonate side chains. We can only speculate the reason for this to be the formation of a more stable transition state during the decomposition reaction by the creation of six-membered rings. Several mixtures of LiTFSI and LiMMnm11 salts were prepared in order to obtain low melting temperature eutectics, and compared to LiTFSI (m.p. 234 °C), the melting points decrease as expected.22
In all systems, the formation of an amorphous phase was observed (Fig. 3) and above 20–30 mol% of LiMMnm11 the systems were fully amorphous. These systems are very phase-stable with no crystallization detectable even after three months, while for 40–50 mol% of LiMM3311 the systems crystallize, likely driven by anion stacking (Fig. S12, ESI†).
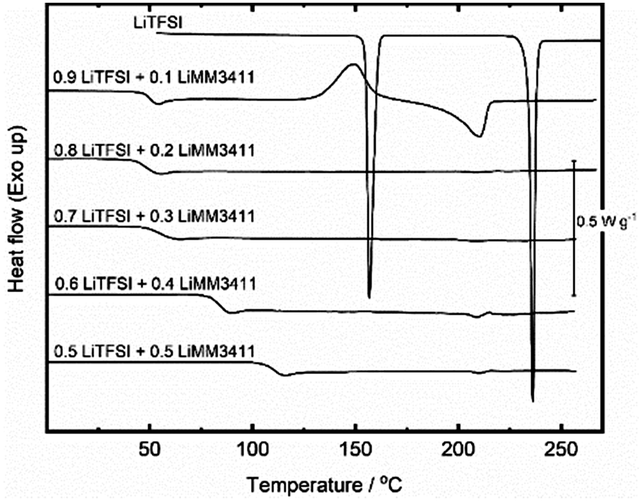 | ||
| Fig. 3 DSC traces of the LiTFSI–LiMM3411 systems with compositions in molar fractions. | ||
Obtaining glassy solids simply by solvent evaporation is surprising as it most often results in salt precipitation as the concentration increases. In contrast, we observe here a rapid increase in viscosity of the liquids during drying, which causes the activation energy for fluid flow to exceed the energy gain either by crystal formation or growth.23 Although this mechanism is often found for quenched salt melts, we believe it applies here when our mixtures lose water. This also explains the increase in the glass transition temperature for the compositions with more MMnm11 anion content, as their aqueous solutions have higher viscosities and lose fluidity at higher temperatures.
The abrupt increase in viscosity prevents the mixtures to get fully dry by hindering the diffusion of water, resulting in residual water content of 0.1–0.2 wt%, ≫ the ca. 100 ppm reported for other solvent-free electrolytes. This is, however, expected to have little/no effect on the electrochemistry given its low mobility and activity, as has recently been established for water-in-salt electrolytes with much higher water contents.24
By mixing LiTFSI with the LiMMnm11 salts significantly decreased melting points were achieved, but the glass transition temperatures are still above 50 °C. This renders them too viscous for battery application, even for HT-LIBs. In order to further decrease the glass transition temperatures and the viscosities, a series of quaternary systems, i.e. Li1−xNax(TFSI)1−y(MMnm11)y were made, making use of the larger and therefore less polarising, and less coordinating Na+ cation.25 From DSC data, three quasi-phase diagrams were created for TFSI contents above 50 mol% (Fig. 4). These mixed anion-mixed cation systems have composition areas where no crystalline phases were observed. In the fully amorphous regions (blue), the partial substitution of Li by Na decreased the glass transition temperatures slightly (e.g. A to B in Fig. 4c). This effect was limited up to 20–30 mol% of Na+, where it favours the formation of crystalline phases above these concentrations. The trend observed for the binary systems extends to any cation composition and increasing the MMnm11 content (e.g. A to C in Fig. 4c) produces a corresponding increase in Tg. The ionic conductivities of Li(TFSI)0.8(MM3411)0.2, Li0.8Na0.2(TFSI)0.8(MM3411)0.2 and Li(TFSI)0.7(MM3411)0.3 all follow the same trend; cation substitution (Li to Na) produces only a two-fold increase, while anion substitution a two order of magnitude increase (Fig. 5a) – the latter is related to the ∼10 °C decrease in Tg as 10 mol% of the anion was substituted. The effect is less evident at higher temperatures as the free volume increases and allows all ions to migrate.
 | ||
| Fig. 4 Quasi-phase diagram for the quaternary mixtures containing MM3311, MM4411, and MM3411. The colours are only for visual aid. The bullets represent data points, hollow mark totally amorphous compositions. Values for LixNa1−xTFSI are from the literature.14 | ||
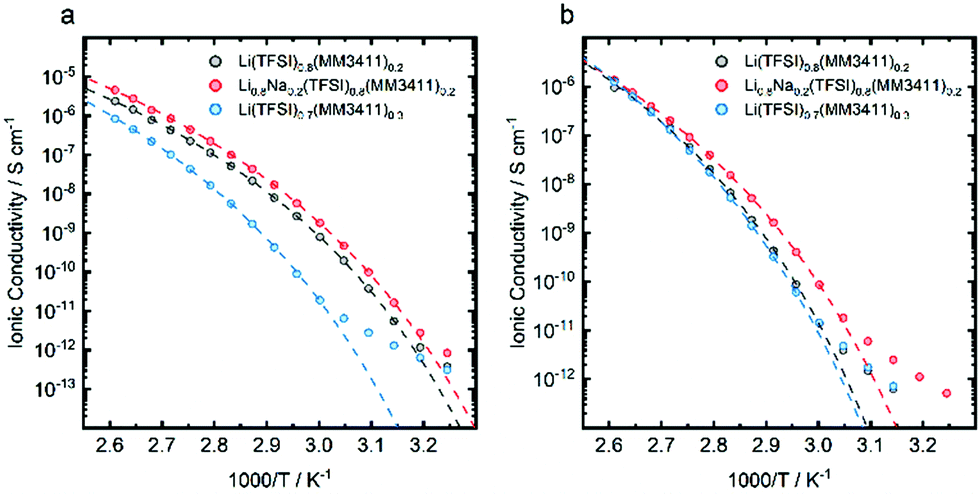 | ||
| Fig. 5 Arrhenius plot of the ionic conductivities of (a) Li(TFSI)0.8(MM3411)0.2, Li0.8Na0.2(TFSI)0.8(MM3411)0.2 and Li(TFSI)0.7(MM3411)0.3, and (b) for mixtures with different MMnm11 anions. | ||
The Tgs obtained from the DSC traces agree with the overall changes in the conductivities observed in the Arrhenius plots; above Tg the ionic conductivity follows the Vogel–Fulcher–Tamman (VFT) relation:
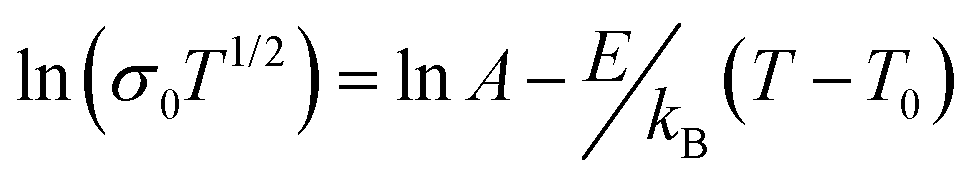 | (1) |
Kubota and Matsumoto31 reported on a more closely related system, with a bulky anion similar to TFSI operating at 150 °C, resulting in a very high Li+ transference number of 0.94. Compared to the system of Kubota (10−2 S cm−1 at 100 °C), the rather low conductivities observed here are likely due to the higher viscosities from our larger anions and a higher degree of ion association.32 Speculatively the sulfonate groups of the MMnm11 anions might act as Li+ traps20 and promote the formation of large aggregates (Fig. 6). The local structure was unfortunately not possible to be unambiguously resolved by e.g. IR or Raman spectroscopy (not shown). The partial substitution of Li+ by Na+ perturbs the aggregate structure, creates free volume, and increase the ion conductivity (Fig. 5).
 | ||
| Fig. 6 Schematic of possible aggregate structures. | ||
The atomic fraction of the cation of interest (Li+ or Na+), which would have an influence on the C-rate capability, is ca. 5% (excl. H), close to the 8% for the system of Kubota. In comparison, polymeric single-ion conductors (SICs), with the same ion conduction mechanism, are often at ca. 1%, while crystalline LISICON is at ca. 40%, but the conduction mechanism differs significantly.
From the above, Li(TFSI)0.8(MM3411)0.2 was selected for proof-of-concept of electrochemical stability tests. In the anodic part of the LSV, a two-step decomposition is observed (Fig. 7). The first step at 3.5 V vs. Li+/Li is attributed to the decomposition of MM3411, expected to have a lower oxidation potential than TFSI,20 while the second step, above 5 V vs. Li+/Li likely corresponds to TFSI decomposition.33 Both steps are hard to resolve in the voltammogram as the current responses overlap. This can be attributed to the high electrolyte viscosity, resulting in slow diffusion of MM-anion decomposition by-products away from the electrode surface. The “tail” in the trace corresponds to a constant anion decomposition at the electrode surface. As compared to liquid LIB electrolytes a reduced slope is observed, simply an effect of viscosity induced mass transfer limitations.
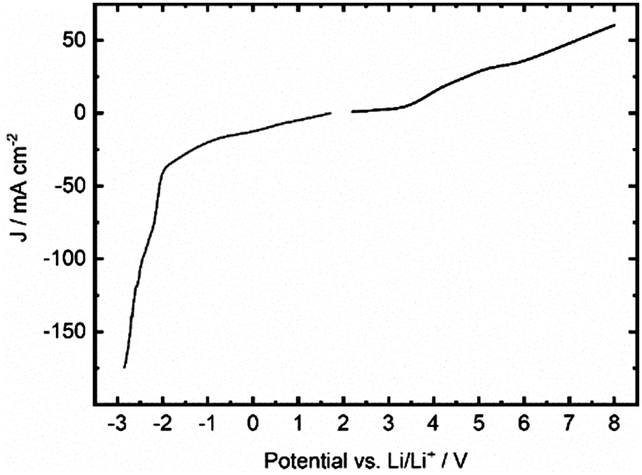 | ||
| Fig. 7 Cathodic and anodic LSV traces for Li(TFSI)0.8(MM3411)0.2. | ||
On the cathodic side, due to the low electrolyte conductivity, the small peak at ca. 0 V vs. Li/Li+ is truly believed to correspond to minor lithium ion reduction and lithium metal plating, while the large feature below −2 V vs. Li/Li+ is due to TFSI anion reduction and decomposition. This very low cathodic limit differs significantly from the reduction potential previously predicted for TFSI,34 likely due to a large polarization build up between working and counter electrodes in the experimental set-up.
In conclusion, high purity and yield synthesis and full characterization of new Li/Na salts based on pseudo-delocalized anions (MMnm11) was reported. Different combinations with LiTFSI were investigated as prospective battery electrolytes. The presence of 20–30 mol% of the MMnm11 anions prevents completely crystallization, which proves the possible preparation at moderate temperatures (ca. 60 °C) of a liquid completely solvent-free electrolytes. The effect is more evident for the asymmetric MM3411 anion as organized packing is further inhibited and opens for further exploring alternative flexible and bulky anions to form solvent-free battery electrolytes. Additionally, the presence of the bulky pseudo-delocalized MMnm11 anions seems to have a positive effect on the Li+ transference number. Apart from using the asymmetry of the MMnm11 anions the ionic conductivity might be improved by reducing the ion–ion interaction further by replacing the –SO3− moieties by more weakly cation coordinating units e.g. –SO2–N−–SO2CF3, as already shown effective in block co-polymer SICs.30
We are grateful for the support by Chalmers, the Erasmus Mundus joint master program; Materials for Energy Storage and Conversion (M.E.S.C.) (JFS) and the financial support by the Swedish Energy Agency (“Batterifonden” grants #37671-1 & #37685-1), as well as Chalmers Battery Initiative and Areas of Advance; Energy, Materials Science, and Transport.
Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Ozawa, Solid State Ionics, 1994, 69, 212–221 CrossRef CAS.
- Y. Matsuda, M. Morita and T. Yamashita, J. Electrochem. Soc., 1984, 131, 2821–2827 CrossRef CAS.
- K. Xu, J. Electrochem. Soc., 2008, 155, A733 CrossRef CAS.
- M. Montanino, S. Passerini and G. B. Appetecchi, Rechargeable Lithium Batteries: From Fundamentals to Applications, Woodhead Publishing, 2015, pp. 73–116 Search PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS PubMed.
- J. Wen, Y. Yu and C. Chen, Mater. Express, 2012, 2, 197–212 CrossRef CAS.
- A. Balducci, Top. Curr. Chem., 2017, 375, 20 CrossRef PubMed.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525–2540 RSC.
- J. G. Kim, B. Son, S. Mukherjee, N. Schuppert, A. Bates, O. Kwon, M. J. Choi, H. Y. Chung and S. Park, J. Power Sources, 2015, 282, 299–322 CrossRef CAS.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 RSC.
- N. Plylahan, M. Kerner, D.-H. Lim, A. Matic and P. Johansson, Electrochim. Acta, 2016, 216, 24–34 CrossRef CAS.
- R. A. Guidotti and P. Masset, J. Power Sources, 2006, 161, 1443–1449 CrossRef CAS.
- K. Kubota, T. Nohira, T. Goto and R. Hagiwara, J. Chem. Eng. Data, 2008, 53, 2144–2147 CrossRef CAS.
- R. Hagiwara, K. Tamaki, K. Kubota, T. Goto and T. Nohira, J. Chem. Eng. Data, 2008, 53, 355–358 CrossRef CAS.
- F. Xu, C. Liu, W. Feng, J. Nie, H. Li, X. Huang and Z. Zhou, Electrochim. Acta, 2014, 135, 217–223 CrossRef CAS.
- X. Tu, Y. Chu and C. Ma, Ionics, 2010, 16, 81–84 CrossRef CAS.
- A. Watarai, K. Kubota, M. Yamagata, T. Goto, T. Nohira, R. Hagiwara, K. Ui and N. Kumagai, J. Power Sources, 2008, 183, 724–729 CrossRef CAS.
- J. O. Isard, J. Non-Cryst. Solids, 1969, 1, 235–261 CrossRef CAS.
- B. Chowdari, R. Huq and G. Farrington, Solid State Ionics, 1992, 57, 49–58 CrossRef CAS.
- E. Jónsson, M. Armand and P. Johansson, Phys. Chem. Chem. Phys., 2012, 14, 6021 RSC.
- E. Hosseini-Bab-Anari, A. Boschin, T. Mandai, H. Masu, K. Moth-Poulsen and P. Johansson, RSC Adv., 2016, 6, 85194–85201 RSC.
- M. Hillert, Phase Equilibria, Phase Diagrams and Phase Transformations, Cambridge University Press, Cambridge, 2nd edn, 2008 Search PubMed.
- D. Turnbull and M. H. Cohen, J. Chem. Phys., 1958, 29, 1049–1054 CrossRef CAS.
- M. S. Ding, A. Von Cresce and K. Xu, J. Phys. Chem. C, 2017, 121, 2149–2153 CrossRef CAS.
- A. Ponrouch, D. Monti, A. Boschin, B. Steen, P. Johansson and M. R. Palacín, J. Mater. Chem. A, 2015, 3, 22–42 RSC.
- D. Bamford, A. Reiche, G. Dlubek, F. Alloin, J.-Y. Sanchez and M. A. Alam, J. Chem. Phys., 2003, 118, 9420–9432 CrossRef CAS.
- G. Dlubek, Y. Yu, R. Krause-Rehberg, W. Beichel, S. Bulut, N. Pogodina, I. Krossing and C. Friedrich, J. Chem. Phys., 2010, 133, 124502 CrossRef CAS PubMed.
- L. S. García-Colín, L. F. del Castillo and P. Goldstein, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 40, 7040–7044 CrossRef.
- A. P. Abbott, ChemPhysChem, 2005, 6, 2502–2505 CrossRef CAS PubMed.
- R. Bouchet, et al. , Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed.
- K. Kubota and H. Matsumoto, J. Phys. Chem. C, 2013, 117, 18829–18836 CrossRef CAS.
- D. R. MacFarlane, M. Forsyth, E. I. Izgorodina, A. P. Abbott, G. Annat and K. Fraser, Phys. Chem. Chem. Phys., 2009, 11, 4962 RSC.
- M. Ue, M. Takehara, S. Mori and M. C. Corporation, J. Electrochem. Soc., 1997, 144, 2684–2688 CrossRef CAS.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938–943 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedure, synthesis of the MMnm11 salts, NMR spectra and additional figures and tables. See DOI: 10.1039/c8cc07076h |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |