
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An in situ combinatorial methodology to synthesize and screen chemical probes†
Antonie J.
van der Zouwen‡
 ,
Jonas
Lohse‡
,
Lianne H. E.
Wieske
,
Katharina F.
Hohmann
,
Ramon
van der Vlag
and
Martin D.
Witte
*
,
Jonas
Lohse‡
,
Lianne H. E.
Wieske
,
Katharina F.
Hohmann
,
Ramon
van der Vlag
and
Martin D.
Witte
*
Chemical Biology II, Stratingh Institute for Chemistry, University of Groningen, 9747AG Groningen, The Netherlands. E-mail: M.D.Witte@rug.nl
First published on 28th January 2019
Abstract
Chemical probes that label proteins of interest in the context of complex biological samples are useful research tools. The reactive group that forms the covalent bond with the target protein has a large effect on the selectivity and selecting the appropriate group determines the success of a probe. We here report the development of a combinatorial methodology based on imine chemistry that enables straightforward in situ synthesis and screening of different reactive groups and thereby simplifies identification of probe leads. Using our methodology, we found chemical probes targeting BirA and chloramphenicol acetyl transferase, two proteins associated with antibacterial activity and resistance.
Chemical probes that covalently introduce a bioorthogonal handle or reporter group onto a protein of interest (POI) have become fundamental research tools.1 They enable functional studies in biologically relevant settings and have been applied to identify the targets of biologically active compounds.2 To prepare chemical probes for a POI, often ligands or inhibitors are functionalized with a reactive group that covalently modifies an amino acid within the proximity of the binding pocket.3 A variety of reactive groups has been employed for this purpose and selecting the appropriate ligand-reactive group combination is key to the success of the probe.4 Not only may the reactive group alter the biological activity, but it also determines to a large extent if proteins get labeled at all.5 Probes will only label their target efficiently if the reactive group is positioned correctly.4k Molecular modeling can address this issue in part, but often knowledge of the targets is lacking and a major challenge therefore remains in finding the optimal ligand-reactive group combination. As a consequence, sets of probes are synthesized that vary in the position and the chemotype of the reactive group.4e,k The synthesis, purification and (bio)chemical analysis of these probes are time consuming and form a serious bottleneck in probe development. Combinatorial approaches and fragment-based screening have been explored to speed-up synthesis of chemical probes.6 However, in the former approach the individual probes still need to be purified and analyzed, while the latter is more suitable for the serendipitous identification of leads. The field of drug discovery has tackled similar challenges by developing combinatorial chemistries that combine preparing and screening of compound libraries in one simple operation.7 Imine chemistry has proven to be suitable for this purpose, since it allows straightforward linking of fragments by mixing reaction partners, with only water as the by-product.
Inspired by these in situ approaches and encouraged by earlier reports of imine-containing probes,8 we set out to develop a similar combinatorial strategy for the synthesis and screening of chemical probes. We here report the first proof-of-concept study of how acylhydrazone and oxime formation can be used to tether ligands and inhibitors to reactive groups in situ (Fig. 1). With this method, we functionalized several α-nucleophiles with commonly used reactive groups. The in situ generated probes label their targets bovine carbonic anhydrase II (CA-II), streptavidin (Strp), avidin (AVI) and BirA. The selectivity and specificity of a particular ligand-reactive group combination is easily assessed by gel-based screening of the reaction mixtures. We believe that our modular approach can dramatically simplify the identification of new leads for probes.
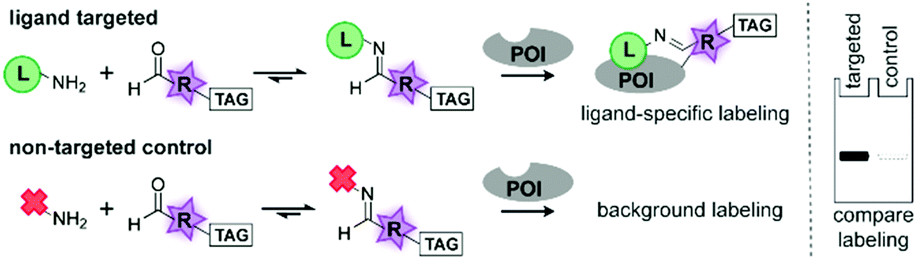 | ||
| Fig. 1 Schematic representation of the combinatorial probe synthesis approach. α-Nucleophile ligands (L) that bind to the protein of interest (POI) are mixed with carbonyl-containing reactive groups (R). The resulting probes modify the POI with a TAG (bioorthogonal handle or fluorophore). By comparing the TAG signal of the targeted probe with a non-targeted control (red cross), the efficiency of the probe can be determined. | ||
To study if combinatorial chemistry can be employed to screen ligand-reactive group combinations, we decided to synthesize aldehyde and ketone derivatives of acyl imidazoles,4a diazotransfer reagents,4b Michael acceptors,4c alkylating agents,4d sulfonyl fluorides,4e–g sulfonate esters,4h Woodward's reagent K4i and photocrosslinkers4j–l (Fig. 2). These reactive groups are commonly used in chemical probes and target a broad panel of amino acids. The required aldehydes R1–R9 were readily prepared from commercially available building blocks in no more than four steps. Considering that R1–R5 could decompose in the presence of α-nucleophiles, we first evaluated if in situ synthesis of the probes was feasible. 1H-NMR (Fig. S1, ESI†) and UPLC-MS (Fig. S2, ESI†) experiments with acethydrazide and methoxyamine revealed that R1–R3 and R5 can be linked to ligands using imine chemistry. However, R4 is not suitable, as it readily decomposed, even in the absence of an α-nucleophile.
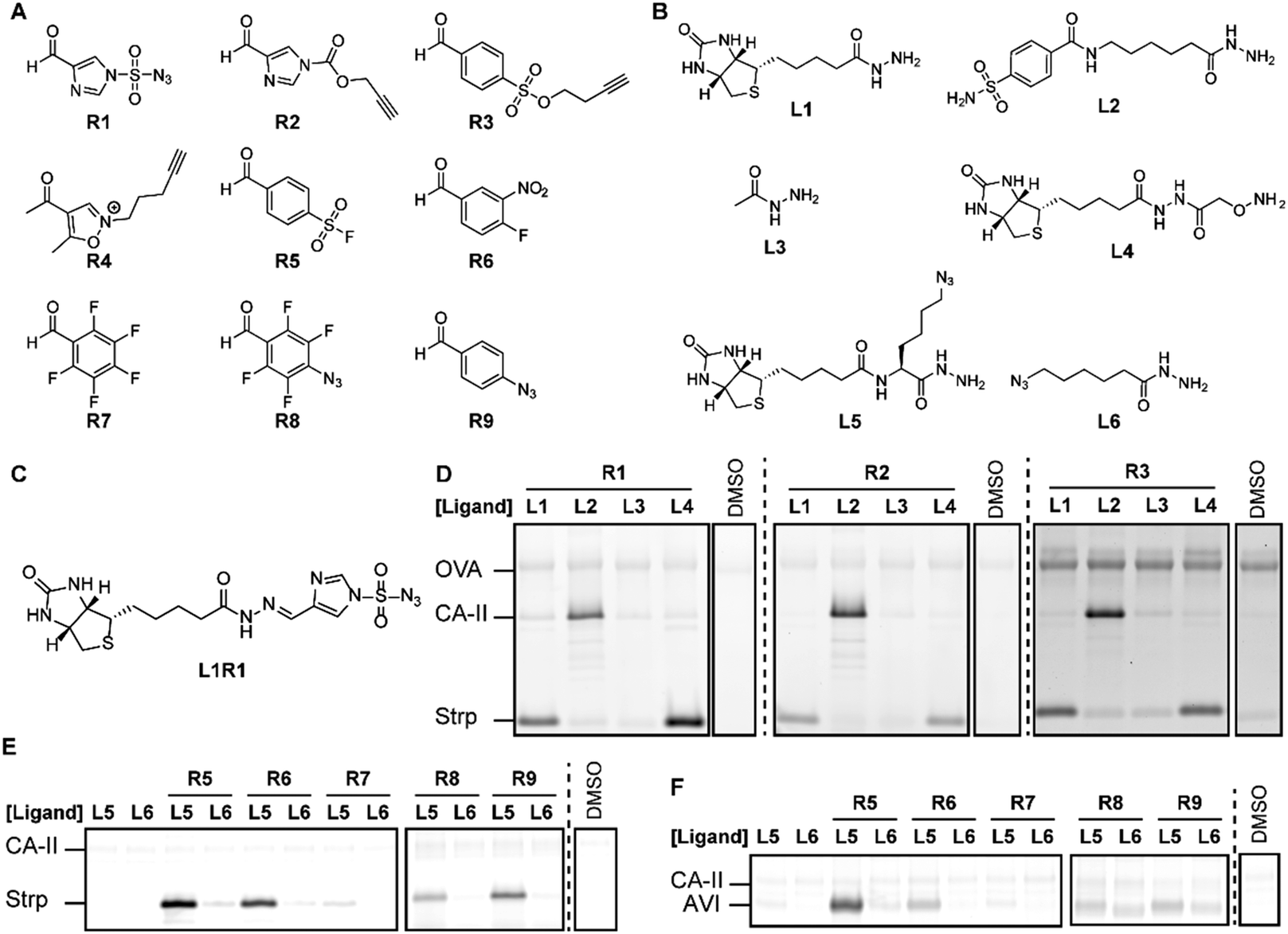 | ||
| Fig. 2 Development and screening of the imine chemical probes. (A) Carbonyl-containing reactive groups. (B) Biotin and benzenesulfonamide α-nucleophile ligands. (C) Example of an in situ generated chemical probe. (D) Probes prepared from ligands L1–L4 and reagents R1–R3 modify their respective targets Strp and CA-II. (E and F) Incorporation of an azido reporter group allows labeling of Strp (E) and avidin (F) using probes prepared with ligands L5–L6 and reagents R5–R9. The DMSO lanes are representative examples of background fluorescence. | ||
Next, we assessed if the in situ formed probes could be used for protein labeling experiments on a mixture of Strp, CA-II and ovalbumin (OVA). We selected this model system, because Strp and CA-II are readily modified with chemical probes that contain a biotin or benzenesulfonamide targeting group, respectively.4h,9 The included OVA serves as a first control for protein specificity. By reacting R1–R4 with an equimolar amount of biotin hydrazide L1, benzenesulfonamide hydrazide L2 or biotin alkoxyamine L4 overnight, we prepared a library of probes that target Strp and CA-II. We also prepared the corresponding non-targeted reagents from acethydrazide L3 as a second control for ligand-independent labeling. For the proof-of-concept studies, we followed previously reported labeling protocols.4a,b,k We added the resulting probes to the protein mixture in HEPES buffer (pH 7.4) and incubated for 2 hours. Subsequently, the bioorthogonal handle introduced by the probe was reacted with a fluorophore reporter group using either copper catalyzed or strain-promoted alkyne–azide cycloaddition reactions. The in-gel fluorescence revealed that incubating the protein mixture with acethydrazide L3-based reagents resulted in minimal labeling of the proteins (Fig. 2D). The fluorescence intensity for Strp or CA-II increased considerably when we reacted the aldehydes R1–R3 either with biotin ligands L1 or L4, or benzenesulfonamide hydrazide L2, respectively. At the same time the signal for OVA was left unchanged, which underlines that the target proteins only get labeled efficiently in the presence of the appropriate ligand. Comparing the fluorescence intensity of the labeled proteins indicated that the in situ formed probes containing R1 or R2 label Strp and CA-II with a similar efficiency. The labeling with R3 is considerably weaker, even when the reaction time was extended to 6 h. As was expected from the NMR studies, we only detected a fluorescent signal for R4 when functionalized with biotin alkoxyamine L4 (Fig. S3, ESI†).
We then performed a similar experiment with R5–R9. These reagents lack an azide or alkyne handle. To detect the labeled proteins, we performed a transimination reaction with an excess of a hydroxylamine fluorophore as has been reported previously.8a Unfortunately, inefficient exchange of the imine hampered reliable detection of protein–probe adducts (Fig. S3, ESI†). We therefore evaluated the suitability of R5–R9 for in situ probe formation on Strp and avidin using azide-functionalized ligands L5 and L6. These biotin-binding proteins only got labeled efficiently when R5–R9 were bound to biotin ligand L5. Besides the ligand, also the reactive group had a large effect on the outcome of the experiment, which underscores the importance of screening different reactive groups. Sulfonyl fluoride L5R5 labeled both target proteins strongly, while aryl fluoride L5R7 modified neither of them (Fig. 2 and Fig. S3, ESI†). The remaining probes did label Strp, but labeled avidin only very weakly, despite the structural similarity between these proteins.
These initial studies demonstrated that in situ probe formation with R1–R3 and R5–R9 is feasible and that incomplete probe formation does not cause off-target labeling. This was further confirmed by extending the incubation time (Fig. S4–S6, ESI†) and by lowering the concentration of the target protein, while keeping the OVA and probe concentrations constant (Fig. S7, ESI†). Neither resulted in increased labeling of the control protein. Even when using multiple probes in a single sample, we observed selective labeling of the targeted proteins (Fig. S8, ESI†), suggesting that our method may be suitable to screen multiple reactive groups in the same sample.
To demonstrate that our modular probe synthesis strategy can be extended to other proteins, we applied it on BirA from E. coli. This essential biotin ligase has emerged as a target for the development of antibacterial agents10 and we recently showed that a biotin-diazotransfer reagent modifies purified BirA.11 Unfortunately, this reagent did not label BirA efficiently in more complex samples. To identify new probe leads, we derivatized biotin ligands L1, L4 and L5 with reactive groups R1–R3 and R5–R9 and incubated cell lysate of BirA-overexpressing E. coli with 22.2 μM of the resulting probes for 2 hours. Analysis of the labeling reaction by fluorescence scanning (for the probes that contain a bioorthogonal handle) or by western blotting (for the probes that attach biotin covalently to their targets) showed that acyl imidazoles L1R2 and L4R2, sulfonyl fluorides L1R5 and L4R5, alkylating agent L5R7 and photocrosslinker L1R8, L4R8 and L5R8 label BirA rather selectively (Fig. 3 and Fig. S9, ESI†). Interestingly, biotin sulfonyl fluoride L5R5 labeled, besides BirA, an additional unidentified protein of approximately 25 kDa. Control probe L6R5 and biotin-based probes L1R5 and L4R5 labeled this off-target only weakly, suggesting that labeling is ligand-dependent. The other probes either did not label BirA or showed the same labeling intensity as the control probes. Again, the reactive group preferences slightly differ from the other biotin-binding proteins. Molecular docking of the probes in the X-ray crystal structure of BirA (PDB ID: 4WF2) provided some insight why acyl imidazole R2, sulfonyl fluoride R5 and alkylating agent R7 are preferred. Several lysine residues are suitably positioned near the entrance of the active site and these might react with these reactive groups (Fig. S10, ESI†). Moreover, the pentafluoroaryl group of L5R7 is in close proximity of a methionine residue that might be alkylated. This residue seems to be out of reach of the other pentafluoroaryl probes.
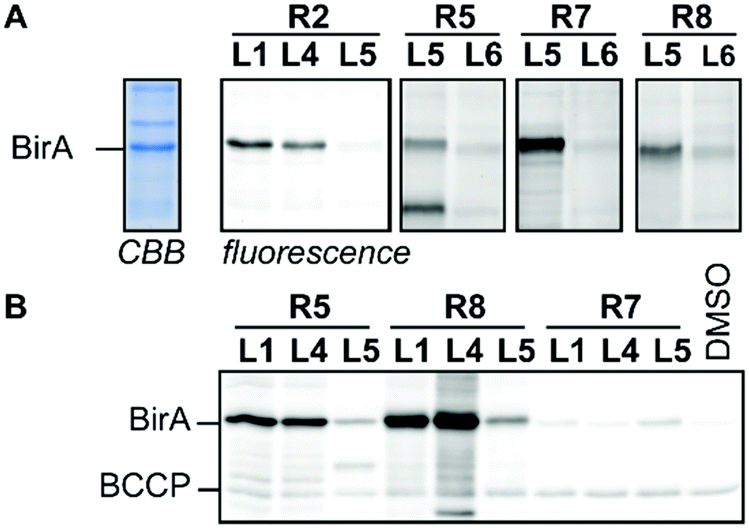 | ||
| Fig. 3 Profiling studies in cell lysate of BirA-overexpressing E. coli. (A) In-gel fluorescence using BODIPY-azide or BODIPY-alkyne as read-out. The CBB lane is a representative example of the total protein content in the cell lysate. (B) Western blotting using streptavidin–horseradish peroxidase as read-out. CBB: Coomassie Brilliant Blue, BCCP: biotin carboxy carrier protein. For full gel images see Fig. S9 (ESI†). | ||
Next, we analyzed the leads of the screen further by varying the incubation time and the amount of probe. For most of the probes, labeling of BirA reached a plateau after 2–4 hours (Fig. S11, ESI†). The labeling efficiency also demonstrated to be concentration dependent. At low concentrations, all probes showed weak labeling of BirA. The signal intensity improved drastically when we increased the probe concentration. However, only L1R5, L4R5, L5R7 and L1R8 labeled cleanly at higher concentrations (Fig. S12, ESI†). Of these probes, we decided to determine the detection limit of L1R5 and L5R7. Even though as little as 10 ng of BirA (275 fmol) can be visualized in E. coli lysate with these probes (Fig. S13, ESI†), BirA could not be detected in native lysates, presumably due to the low copy number of BirA per cell (Fig. S14, ESI†).12 Nonetheless, these leads form a good starting point for the further development of more sensitive probes.
Finally, we focused our attention on the off-target of L5R5. Labeling of this protein is concentration- and time-dependent and could be abolished by heat-denaturing the protein (Fig. 4A, B and Fig. S15, ESI†). Chemoproteomic analysis of the labeled protein identified chloramphenicol acetyl transferase 3 (CAT) as the major hit (Fig. S15 and Table S2, ESI†). The codon-optimized E. coli strain that we used for the profiling studies carries a pACYC-based plasmid that codes for the expression of this enzyme. Competition experiments with CAT substrate chloramphenicol or known inhibitor fusidic acid confirmed that L5R5 indeed labeled CAT (Fig. 4C). Both compounds diminished the signal in a concentration-dependent fashion, which suggests that L5R5 binds in the same binding pocket. A recent study demonstrated that CAT also can be labeled with pentafluoroaryl reactive groups similar to R7,13 but in our case we only observed good labeling when using sulfonyl fluoride L5R5. Interestingly, sulfonyl fluoride probes based on L1 and L4 did not. Furthermore, competition with biotin did not block labeling of CAT, indicating that the azidolysine is important for the targeting of CAT. To validate this hypothesis and to target CAT selectively over BirA, we synthesized benzoyl- and butyryl-azidolysine ligands L7 and L8. Furthermore, we also prepared azido-D-serine hydrazide L9 as a chloramphenicol mimic. Of the newly designed probes, only L7R5 and L8R5 labeled CAT (Fig. 4D and Fig. S15, ESI†). As expected, replacing the biotin for a benzoyl or butyryl group leads to reduced BirA labeling. Docking of the probes in the X-ray crystal structure of CAT bound to fusidic acid (PDB ID: 1Q23) showed that they may adopt a pose in which the sulfonyl fluoride is in proximity to catalytic histidine residue 193 (Fig. S16, ESI†). Alkylation of this histidine with methyl 4-nitrobenzenesulfonate14 inhibited labeling of CAT, indicating that this residue might be modified by our probes. However, several of the sulfonyl fluorides that did not label CAT, including L9R5, adopt a similar pose. While the docking helps to explain the outcome, its predictive value is limited, once more underpinning the importance of screening combinations.
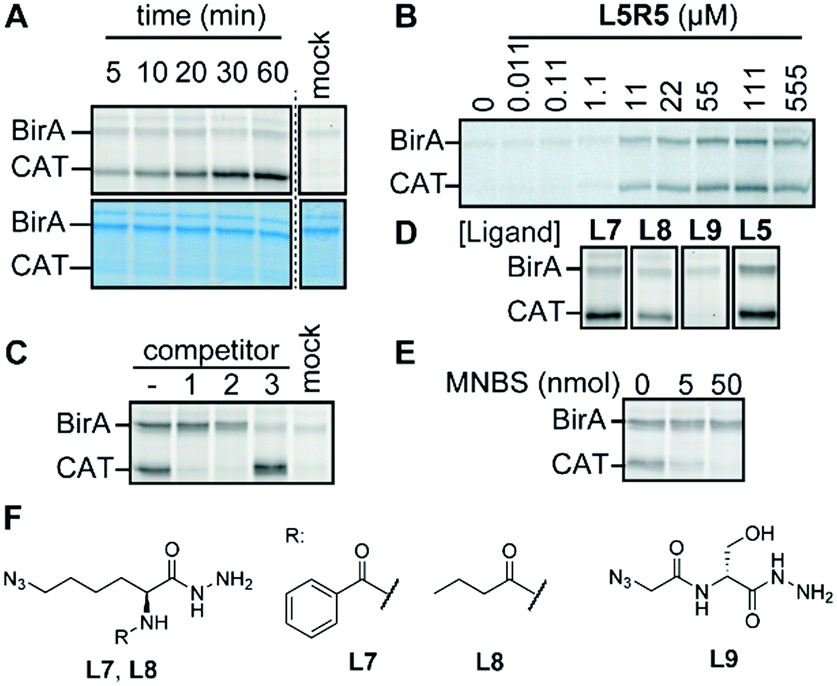 | ||
| Fig. 4 (A) Time dependency of CAT labeling by L5R5 in the presence of 10 μM biotin. (B) Concentration dependency of BirA and CAT labeling by L5R5. (C) Competition experiments with L5R5. Competitors: (−) no competitor, (1) chloramphenicol (10 nmol), (2) fusidic acid (10 nmol), (3) biotin (50 nmol). (D) Labeling of CAT and BirA with L7R5, L8R5, L9R5 and L5R5. (E) Competition experiment between methyl 4-nitrobenzenesulfonate (MNBS) and L5R5. (F) Structures of L7–L9. All samples in A, C–E contain 200 pmol probe, except the mock, which does not contain probe. Typical sample size: 9 μL. | ||
In conclusion, we here describe a novel combinatorial approach to prepare chemical probes. Readily available carbonyl-functionalized protein labeling reagents can be reacted with α-nucleophiles to form chemical probes that modify their targets selectively within a mixture of proteins and cell lysates. The described methodology allows straightforward screening of the selectivity of ligand-reactive group combinations using reaction mixtures. The identification of novel probes for CA-II and three different biotin-binding proteins, and the serendipitous discovery of a probe for CAT underlines the power of our methodology. Hydrazides are readily prepared from acids or esters. Therefore, a large number of different scaffolds can be converted into suitable ligands for our methodology. As a consequence, our methodology should be applicable towards many different protein targets. Thus, combined with the simplified development of probes and the power of straightforward screening of reactive groups, we envision that our modular probe synthesis methodology will considerably simplify chemical probe development.
The authors thank N. Danda and C. P. Williams for producing lysates of BirA-overexpressing E. coli and A. K. H. Hirsch for the useful discussions and support with docking. The Netherlands Organisation for Scientific Research (NWO-VIDI) grant number 723.015.004 (M. D. W., A. J. V. D. Z.); 723.014.008 (R. V. D. V.) and the Marie Sklowdoska Curie-CIG (CIG PCIG14-GA-2013-631057 to M. D. W.) are acknowledged for funding.
Conflicts of interest
There are no conflicts of interest to declare.References
- (a) M. J. Niphakis and B. F. Cravatt, Annu. Rev. Biochem., 2014, 83, 341–377 CrossRef CAS PubMed; (b) R. Kubota and I. Hamachi, Chem. Soc. Rev., 2015, 44, 4454–4471 RSC; (c) M. Garland, J. J. Yim and M. Bogyo, Cell Chem. Biol., 2016, 23, 122–136 CrossRef CAS PubMed.
- S. Pan, H. Zhang, C. Wang, S. C. L. Yao and S. Q. Yao, Nat. Prod. Rep., 2016, 33, 612–620 RSC.
- (a) M. J. C. Long, J. R. Poganik and Y. Aye, J. Am. Chem. Soc., 2016, 138, 3610–3622 CrossRef CAS PubMed; (b) K. Amaike, T. Tamura and I. Hamachi, Chem. Commun., 2017, 53, 11972–11983 RSC.
- (a) S. Fujishima, R. Yasui, T. Miki, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 3961–3964 CrossRef CAS PubMed; (b) J. Lohse, L. J. Y. M. Swier, R. C. Oudshoorn, G. Médard, B. Kuster, D. J. Slotboom and M. D. Witte, Bioconjugate Chem., 2017, 28, 913–917 CrossRef CAS PubMed; (c) J. Ge, C.-J. Zhang, L. Li, L. M. Chong, X. Wu, P. Hao, S. K. Sze and S. Q. Yao, ACS Chem. Biol., 2013, 8, 2577–2585 CrossRef CAS PubMed; (d) L. M. McGregor, M. L. Jenkins, C. Kerwin, J. E. Burke and K. M. Shokat, Biochemistry, 2017, 56, 3178–3183 CrossRef CAS PubMed; (e) A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC; (f) Q. Zhao, X. Ouyang, X. Wan, K. S. Gajiwala, J. C. Kath, L. H. Jones, A. L. Burlingame and J. Taunton, J. Am. Chem. Soc., 2017, 139, 680–685 CrossRef CAS PubMed; (g) O. Fadeyi, M. D. Parikh, M. Z. Chen, R. E. Kyne, A. P. Taylor, I. O’Doherty, S. E. Kaiser, S. Barbas, S. Niessen, M. Shi, S. L. Weinrich, J. C. Kath, L. H. Jones and R. P. Robinson, ChemBioChem, 2016, 17, 1925–1930 CrossRef CAS PubMed; (h) S. Tsukiji, M. Miyagawa, Y. Takaoka, T. Tamura and I. Hamachi, Nat. Chem. Biol., 2009, 5, 341–343 CrossRef CAS PubMed; (i) P. Martín-Gago, E. K. Fansa, M. Winzker, S. Murarka, P. Janning, C. Schultz-Fademrecht, M. Baumann, A. Wittinghofer and H. Waldmann, Cell Chem. Biol., 2017, 24, 589–597 CrossRef PubMed; (j) Z. Li, D. Wang, L. Li, S. Pan, Z. Na, C. Y. J. Tan and S. Q. Yao, J. Am. Chem. Soc., 2014, 136, 9990–9998 CrossRef CAS PubMed; (k) M. H. Wright, C. Fetzer and S. A. Sieber, J. Am. Chem. Soc., 2017, 139, 6152–6159 CrossRef CAS PubMed; (l) H. Park, J. Y. Koo, Y. V. V. Srikanth, S. Oh, J. Lee, J. Park and S. B. Park, Chem. Commun., 2016, 52, 5828–5831 RSC.
- (a) A. Borodovsky, H. Ovaa, N. Kolli, T. Gan-Erdene, K. D. Wilkinson, H. L. Ploegh and B. M. Kessler, Chem. Biol., 2002, 9, 1149–1159 CrossRef CAS PubMed; (b) S. M. Hacker, K. M. Backus, M. R. Lazear, S. Forli, B. E. Correia and B. F. Cravatt, Nat. Chem., 2017, 9, 1181–1190 CrossRef CAS PubMed.
- (a) J. Ge, X. Cheng, L. P. Tan and S. Q. Yao, Chem. Commun., 2012, 48, 4453 RSC; (b) J. T. Bush, L. J. Walport, J. F. McGouran, I. K. H. Leung, G. Berridge, S. S. van Berkel, A. Basak, B. M. Kessler and C. J. Schofield, Chem. Sci., 2013, 4, 4115 RSC; (c) C. G. Parker, A. Galmozzi, Y. Wang, B. E. Correia, K. Sasaki, C. M. Joslyn, A. S. Kim, C. L. Cavallaro, R. M. Lawrence, S. R. Johnson, I. Narvaiza, E. Saez and B. F. Cravatt, Cell, 2017, 168, 527–541 CrossRef CAS PubMed.
- M. Mondal and A. K. H. Hirsch, Chem. Soc. Rev., 2015, 44, 2455–2488 RSC.
- (a) Y. Takaoka, H. Tsutsumi, N. Kasagi, E. Nakata and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 3273–3280 CrossRef CAS PubMed; (b) H. Wakabayashi, M. Miyagawa, Y. Koshi, Y. Takaoka, S. Tsukiji and I. Hamachi, Chem. – Asian J., 2008, 3, 1134–1139 CrossRef CAS PubMed.
- L. N. Anderson, P. K. Koech, A. E. Plymale, E. V. Landorf, A. Konopka, F. R. Collart, M. S. Lipton, M. F. Romine and A. T. Wright, ACS Chem. Biol., 2016, 11, 345–354 CrossRef CAS PubMed.
- B. P. Duckworth, T. W. Geders, D. Tiwari, H. I. Boshoff, P. A. Sibbald, C. E. Barry, D. Schnappinger, B. C. Finzel and C. C. Aldrich, Chem. Biol., 2011, 18, 1432–1441 CrossRef CAS PubMed.
- J. Lohse, A. Schindl, N. Danda, C. P. Williams, K. Kramer, B. Kuster, M. D. Witte and G. Médard, Chem. Commun., 2017, 53, 11929–11932 RSC.
- (a) A. M. Femino, F. S. Fay, K. Fogarty, R. H. Singer, X. S. Xie, J. Hearn, A. Emili and X. S. Xie, Science, 1998, 280, 585–590 CrossRef CAS PubMed; (b) G.-W. Li, D. Burkhardt, C. Gross and J. S. Weissman, Cell, 2014, 157, 624–635 CrossRef CAS PubMed.
- A. M. Embaby, S. Schoffelen, C. Kofoed, M. Meldal and F. Diness, Angew. Chem., Int. Ed., 2018, 57, 8022–8026 CrossRef CAS PubMed.
- A. Lewendon and W. V. Shaw, Biochem. J., 1993, 290, 15–19 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures and tables, experimental procedures and compound characterization. See DOI: 10.1039/c8cc06991c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |